
Abstract
Purpose
The diagnostic rate for Mendelian diseases by exome sequencing (ES) is typically 20–40%. The low rate is partly because ES misses deep-intronic or synonymous variants leading to aberrant splicing. In this study, we aimed to apply RNA sequencing (RNA-seq) to efficiently detect the aberrant splicings and their related variants.
Methods
Aberrant splicing in biopsied muscles from six nemaline myopathy (NM) cases unresolved by ES were analyzed with RNA-seq. Variants related to detected aberrant splicing events were analyzed with Sanger sequencing. Detected variants were screened in NM patients unresolved by ES.
Results
We identified a novel deep-intronic NEB pathogenic variant, c.1569+339A>G in one case, and another novel synonymous NEB pathogenic variant, c.24684G>C (p.Ser8228Ser) in three cases. The c.24684G>C variant was observed to be the most frequent among all NEB pathogenic variants in normal Japanese populations with a frequency of 1 in 178 (20 alleles in 3552 individuals), but was previously unrecognized. Expanded screening of the variant identified it in a further four previously unsolved nemaline myopathy cases.
Conclusion
These results indicated that RNA-seq may be able to solve a large proportion of previously undiagnosed muscle diseases.
Similar content being viewed by others

INTRODUCTION
Currently, exome sequencing (ES) is a first-line approach for Mendelian disease diagnosis.1,2,3 Though ES comprehensively covers exons and the flanking introns of protein-coding genes, its diagnosis rate is not high (20–40%) (refs. 3,4,5,6). One reason for such low diagnostic rates is that ES overlooks the synonymous variants leading to aberrant splicing. Though ES can detect synonymous variants, it is practically difficult to evaluate the pathogenicity of numerous synonymous variants. In addition, ES does not cover deep-intronic regions where variants causing aberrant splicing may exist.7,8,9
To address these problems, RNA sequencing (RNA-seq) is a reasonable next choice. Theoretically, RNA-seq can directly detect ectopic splicing events in transcripts, followed by detecting causative variants of the aberrant splicing events in the genome. Previously, two groups independently developed workflows and applied them to patients’ samples and identified causative variants.10,11
In this study, we used RNA-seq to diagnose muscle disease cases previously unsolved by ES. We establish a new RNA-seq workflow for detecting possible aberrant splicing events, which can produce reasonable numbers of candidate variants. By applying the workflow to unsolved cases of nemaline myopathy (NM [MIM 256030]), new pathogenic variants associated with aberrant splicing are identified and described.
MATERIALS AND METHODS
Samples
The study was approved by the institutional review board of Yokohama City University School of Medicine (Yokohama, Japan) and National Center of Neurology and Psychiatry (Kodaira, Japan). We obtained informed consent from the patients or their parents.
RNA-seq
Total RNA was extracted from frozen skeletal muscles (SkM) with the RNeasy Fibrous Tissue Mini Kit (Qiagen, Hilden, Germany), and from fibroblasts (Fb) and lymphoblastoid cell lines (LCL) with the RNeasy Mini Kit (Qiagen) following the manufacturer’s instructions. RNA integrity number of total RNA was more than seven in all samples. From total RNA, poly(A) RNA was selected and the complementary DNA (cDNA) libraries were prepared with the SureSelect Strand-Specific RNA Library Prep Kit (Agilent Technologies, Santa Clara, CA, USA) following the manufacturer’s instructions. The cDNA libraries were sequenced on an Illumina HiSeq 2000 system (Illumina, San Diego, CA, USA) using 92-bp paired-end reads. The generated FASTQ were mapped with STAR 2.5.2b software using the twopassMode function following the software instruction manual.12 Genome index files were generated using the human reference genome (hg19) and the University of California–Santa Cruz (UCSC) gene annotation file (hg19). We visualized the aberrant splicing events using Sashimi plots generated with MISO 0.5.3 (ref. 13).
Principal component analysis (PCA)
We normalized the data considering library sizes and carried out log transformation of the read counts of each gene with the varianceStabilizingTransformation function of the DESeq2 package 1.14.1 of R software.14,15 PCA was performed with the prcomp function of the stats package of R. Six SkM, 2 Fb, and 12 LCL samples were analyzed.
Downsampling analysis of coverage of splicing junctions
Coverage of splicing junctions in 161 genes associated with muscle diseases was analyzed. The 161 genes were extracted from ten categories within the online GeneTable of Neuromuscular Disorders (http://www.musclegenetable.fr): congenital myopathies, distal myopathies, other myopathies, myotonic syndromes, ion channel muscle diseases, malignant hyperthermia, metabolic myopathies, muscular dystrophies, congenital muscular dystrophies, and other neuromuscular disorders. Based on cDNA references of the 161 genes used in Human Gene Mutation Database (HGMD), splicing junctions were extracted. As the counts of reads spanning the splicing junctions and the counts of uniquely mapped reads decreased proportionally, we analyzed the proportion of the splicing junctions with 100 or more reads over the all splicing junctions in the 161 genes. The same samples described in the PCA section were analyzed.
Differential splicing analysis
To quantitate each splicing event, we calculated 5′ or 3′ ψ of each splicing.16 Then, we obtained the difference in 5′ or 3′ ψ (ψ-D) and the fold change of 5′ or 3′ ψ (ψ-FC) for each splicing event in each case of interest compared with four cases with undiagnosed myopathy of nonspecific muscle pathology (NP cases) as reference. To analyze statistical significance of difference in quantity of each splicing event, we used LeafCutter,17 a software calculating p value based on Dirichlet multinomial generalized linear model as previously described by Kremer et al.10 We changed the option arguments following the instructions in the software manual to cover genes with low expression. In particular, we decreased the threshold for the minimum number of reads as follows: m (MINCLUREADS): 8 for intron clustering analysis and c (MIN_COVERAGE): 8 for DS analysis. Furthermore, we decreased the threshold for the minimum number of samples in which each splicing event must be: i (MIN_SAMPLES_PER_INTRON): 1, g (MIN_SAMPLES_PER_GROUP): 1 for differential splicing analysis. Note that we did not correct the p value for multiple testing or set the p value threshold as previously described10 because we found that these adjustments missed most of the known aberrant splicing events (see Discussion).
ES and Sanger sequencing
We performed ES and Sanger sequencing as described in our previous publication.18 DNA was extracted from patients’ blood leukocytes with the QuickGene-610L system (Tokyo, Japan, Fujifilm) following the manufacturer’s instructions, or from frozen skeletal muscle using a conventional phenol chloroform extraction method. DNA was captured with the SureSelect Human All Exon V5 or V6 Kit (Agilent).
Allele-specific expression (ASE) analysis
We extracted the heterozygous single-nucleotide variants (SNVs) with Genome Analysis Toolkit (GATK) SelectVariants and counted the RNA-seq reads of the wild-type or altered alleles at each SNV site with GATK ASEReadCounter. The statistical significance of the ratio of the altered allele to each wile-type allele was analyzed with DESeq2 as previously described.10,11 The threshold of the p value for statistical significance was adjusted using Bonferroni correction based on the number of analyzed NEB SNVs in each sample.
Code availability
Codes for RNA-seq analyses in this study are available on request.
RESULTS
First, we sought to determine which tissue was appropriate for RNA-seq diagnosis of muscle diseases and analyzed the following clinically accessible tissues: SkM, Fb, and LCL. PCA showed that samples of the same tissue type formed separate clusters (Fig. 1a). We next evaluated how many genes associated with muscle diseases are expressed in SkM, Fb, and LCL. We analyzed the median of transcripts per million (TPM) of 161 genes related to muscle diseases in SkM, Fb, and LCL. A large proportion of genes had low median of TPM (<10) in Fb (42.9%) and LCL (55.3%) but not in SkM (23.6%) (Fig. 1b). We next evaluated how many of the alternative splicing events in the 161 genes associated with muscle diseases can be assessed in SkM, Fb, and LCL. We performed a downsampling analysis to evaluate the coverage of splicing junctions in the 161 genes (Fig. 1c). By analyzing the relationship between the counts of uniquely mapped reads (x-axis in Fig. 1c) and the proportion of the “analyzable” splicing junctions (splicing junctions with 100 or more reads over the all splicing junctions in the 161 genes; y-axis in Fig. 1c), we found that the coverage of the 161 genes was higher in the SkM samples compared with Fb and LCL, in all ranges of the mapped read counts (Fig. 1c). Thus, we concluded that SkM samples were the most appropriate for investigating aberrant splicing of muscle disease genes with RNA-seq except for a few disease categories (such as congenital muscular dystrophy and myotonic syndrome, which can be analyzed using Fb) (Fig. S1 and S2).

Establishment of RNA-seq analytic workflow. (a) Principal component analysis (PCA) of gene expression in skeletal muscle (SkM), fibroblast (Fb), and lymphoblastoid cell line (LCL) samples. X-axis: principal component 1 (PC1); Y-axis: PC2. (b) Violin plot of median transcripts per million (TPM) of the 161 muscle disease–associated genes. The same samples in (a) were analyzed. Median TPM of each gene in each tissue was plotted. (c) Downsampling analysis of the coverage of splicing junctions for the 161 muscle disease–associated genes. The proportion of splicing junctions with a depth of 100 or more reads in all splicing junctions was calculated. The same samples in (a) were analyzed. Circle: raw data of each sample; line: downsampled data. (d) Calculation of the difference in 5′ or 3′ ψ (ψ-D) and the fold change of 5′ or 3′ ψ (ψ-FC). The scheme depicts a representative example of an aberrant splicing event. The table at the right side the scheme depicts how to calculate the ψ-D and ψ-FC of each splicing event in cases of interest. Prior to these calculations, we calculated the mean number of read counts of each splicing event in four undiagnosed myopathy of nonspecific muscle pathology (NP) cases and used this number as the reference read count. To calculate the ψ of a splicing event (red arc, hereafter “focused event”), we carried out the following steps. (1) We summed the read counts of all splicing events with the same 5′ junction as the focused event in the reference (100 + 2 in this example, hereafter “total read count”). (2) We performed the same calculation for the 3′ junction in the reference. (3) We selected the 5′ or 3′ junction with a higher total read count in the reference (hereafter “selected junction”; in this example, the 5′ junction had a higher total read count (102 > 2) and was the selected junction). (4) We divided the read count of the focused event by the total read count for the selected junction in the reference (2/102, hereafter “ψRef”). (5) We performed the same calculation for the case of interest (20/70, hereafter “ψCoi”). (6) We calculated the ψ-D and ψ-FC of the focused event in the case of interest as follows: ψ-D = ψCoi − ψRef (0.29 − 0.02); ψ-FC = ψCoi/ψRef (0.29/0.02). Arc, splicing event; red arc, focused splicing event as explained in the table; number on arc, number of reads subjected to splicing; Ex, exon; horizontal line, intron; Ref, reference (the mean of four NP cases); Coi, case of interest. (e) Plot of the ψ-D, ψ-FC, and p-values for narrowing down the candidate aberrant splicing events. The three parameters, ψ-D, ψ-FC, and p-value of LeafCutter of eight known aberrant splicing events and the remaining 282,552 events in seven cases with known splice-site pathogenic variants (five Duchenne muscular dystrophy [DMD] and two Ullrich congenital muscular dystrophy [UCMD] cases) (Table S1) were plotted. Note that five known aberrant splicing events were plotted at the same position (arrow). Most of the 282,552 events were assumed to be benign. Values <0 for ψ-D were plotted at zero. Values >100 for ψ-FC were plotted at 100. The right graph depicts the left graph rotated clockwise by 90°. Red circle: aberrant splicing event caused by known splice-site pathogenic variant; gray circle: benign splicing event. (f) Box plot of the number of candidate aberrant splicing events in the seven cases analyzed in (e) after narrowing the number down based on ψ-D, ψ-FC, and p values. The number was counted for all protein-coding genes (All genes) and the 161 causative genes for genetic muscle diseases (MD genes).
Next, we developed an RNA-seq analytic workflow to narrow down candidate aberrant splicing events using SkM samples with confirmed pathogenic splice-site variants. To detect differential splicing, we calculated 5′ or 3′ ψ, previously proposed metrics quantifying the proportion of splicing events in all events sharing the same junctions (Fig. 1d).16 We obtained the difference (ψ-D) and the fold change (ψ-FC) of 5′ or 3′ ψ of each splicing event in each case with confirmed splice-site pathogenic variant(s) compared with the four NP reference cases (Fig. 1d). Furthermore, we obtained the p value for difference in proportions of each splicing event between each case of interest and the four NP reference cases using LeafCutter, a software that calculates p value based on the Dirichlet multinomial generalized linear model. We did not correct the p value for multiple testing because we here used the p value just to remove splicing events of not enough reads. To determine the thresholds for the p value, ψ-D, and ψ-FC, we used these three values of eight confirmed aberrant splicing events as the positive control events and the remaining (normal) 282,552 events as negative control events in the seven cases with confirmed pathogenic splice-site variants: five cases of Duchenne muscular dystrophy (DMD) caused by DMD variants19 and two cases of Ullrich congenital muscular dystrophy (UCMD) caused by COL6A2 variants20 (Table S1 and Fig. 1d). We determined the thresholds as follows: 0.05 for the p value, 0.1 for ψ-D, and 20 for ψ-FC, which did not miss any of the eight confirmed aberrant splicing events.
Next, we checked the number of candidate aberrant splicing events in the above seven samples after filtering based on the selected thresholds. The median number of candidate aberrant splicing events including those caused by known pathogenic splice-site variants (Table S1) was 1 in the 161 genes associated with neuromuscular disorders and 33 across all human genes (Fig. 1e). Therefore, our RNA-seq analytic workflow can reduce the number of candidate aberrant splicing events to a tractable level, without missing any positive control pathogenic events.
We applied this workflow to six genetically undiagnosed NM cases (NM-U) in whom ES identified only a monoallelic likely pathogenic variant in NEB, of which biallelic pathogenic variants are known to cause NM (Table 1) (ref. 21). Before performing RNA-seq, we reanalyzed ES data of NM-U1 to NM-U6 focusing on exonic and near-exon intronic regions. ES identified only one pathogenic variant in each of the cases NM-U1 to NM-U6 (Table 1). These variants were confirmed by Sanger sequencing. All the variants except for c.23029del (p.Tyr7677Metfs*10) were absent from the National Heart, Lung, and Blood Institute Grand Opportunity (NHLBI GO) Exome Sequencing Project (ESP) 6500, gnomAD, Tohoku Medical Megabank Organization (ToMMo) 3.5K Japanese, and the Human Genetic Variation Database (HGVD) (hereafter, variant databases of normal populations), or the HGMD and ClinVar (hereafter, disease-related variant databases).22,23,24 NM-U4 was previously sequenced (corresponding to case 69 in their paper) and reported to harbor the pathogenic variant (Table 1) (refs. 25,26).
The pathogenicity of the canonical splice-site and splice-region variants c.20158–6A>G in NM-U2 and c.7228–2A>G in NM-U6 was supported in silico with ESEfinder 3.0, NetGene2, and NNSPLICE 0.9, and experimentally validated by reverse transcription polymerase chain reaction (RT-PCR) (Fig. S3, S2A, and S2B).27,28,29 In silico software tools predicted that c.20158–6A>G created a novel splicing donor site and a 5-bp exon extension leading to an out-of-frame variant in NM-U2, while c.7228–2A>G resulted in the loss of the wild-type splicing acceptor site in NM-U6 (Fig. S3). RT-PCR confirmed the 5-bp exon extension in NM-U2 (Fig. S4A) and the in-frame deletion (p.Asn2410_Asp2477del) caused by exon 54 skipping in NM-U6 (Fig. S4B).
To confirm the appropriateness of the clinicopathological diagnosis of the NM-U cases from overall transcriptional profiles, we performed PCA on RNA-seq data from SkM samples from the six NP, five DMD, two UCMD, two genetically diagnosed NM with biallelic truncating NEB variants (NM-D), and six NM-U cases. PCA indicated that the NP, NM-D, and NM-U samples except one NP, one NM-D, and NM-U5 were separated from the DMD and UCMD samples (Fig. 2a). This is consistent with their muscle pathology: DMD and UCMD show replacement of fibrous and adipose tissues from muscle fibers while NM usually does not. Thus, the NM-U cases, especially those in the cluster of NP, NM-D, and NM-U cases, likely have an additional pathogenic NEB variant missed by ES, such as either a deep-intronic or synonymous NEB variant leading to aberrant splicing. Such aberrant splicing would be identifiable by RT-PCR, in cases of other genes. However, it is not practical for the NEB gene, because this gene is among the largest genes in the human genome comprising 183 exons with extremely complicated alternative splicing patterns.30 Therefore, our RNA-seq-based comprehensive analytic workflow can be the best way to resolve the NM-U cases with a missing pathogenic allele.

Candidate aberrant splicing events detected with RNA-seq analysis of undiagnosed cases. (a) Quality check of skeletal muscle (SkM) samples by principal component analysis (PCA) of gene expression level. Six undiagnosed myopathy of nonspecific muscle pathology (NP), five Duchenne muscular dystrophy (DMD), two Ullrich congenital muscular dystrophy (UCMD), two genetically diagnosed NM with biallelic truncating NEB variants (NM-D), and six undiagnosed NM (NM-U) cases were analyzed. (b,c) Sashimi plots of RNA-seq reads in NM-U cases: (b) NM-U1 and (c) NM-U2, NM-U3, and NM-U4 . Upper black plot: an NP case; lower red plots: NM-U cases. Variants identified by Sanger sequencing are indicated below the plots. Exonic regions are indicated as solid horizontal lines above the plots. Exon and intron sizes are arbitrary for clear visualization of splicing events. Sample names are described in the upper right or left. The read counts spanning each splicing junction are described on or nearby the corresponding arcs. Arcs indicate splicing junctions, and arcs with asterisks indicate candidate aberrant splicing events after narrowing down the list of candidates based on their ψ-D, ψ-FC, and p values. Numbers on arcs describe the read counts supporting the splicing junctions. Arrows indicate partial intron 175 retention. X- and y-axes indicate corresponding genomic region, and reads per kilobase million (RPKM), respectively. The right-hand plots are an enlarged selection of the left-hand plots in (c). (d) Integrative Genomics Viewer (IGV) image of subsampled RNA-seq reads of the 11-bp region around the c.24684G>C variant in NM-U2. The pink lines indicate each RNA-seq read mapped to the targeted region in the gray background. Reads restricted to exon 175 indicate splicing events excising intron 175, while reads spanning exon 175 and intron 175 indicate intron 175 retention. Exon 175 and intron 175 regions are shown as horizontal solid and dotted lines on top. The c.24684 base is indicated at the bottom. The c.24684G>C variant in each read is colored brown (line). Note that most of the spliced reads did not contain c.24684G>C, while most of the reads retaining intron 175 did contain c.24684G>C. Except for the third base, the first five bases of intron 175 base (c.24684+1 to +5) were the same as those of exon 176 (c.24685 to 24689), implying that reads in line with the dashed vertical line could be erroneously mapped. Differently colored sites in the pink line: bases not matched to the reference sequence.
We applied our RNA-seq analytic workflow to NM-U1–NM-U6. RNA-seq identified 83, 65, 37, 39, 78, and 17 aberrant splicing candidates in NM-U1 to NM-U6, respectively. Among them, 2, 3, 2, 2, 0, and 1 candidates were at NEB.
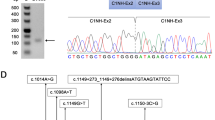
In NM-U1, the two events in NEB involved intronic splice gain leading to the 67-bp novel exon in intron 17 (Fig. 2b). RT-PCR of exons 13 to 21 showed an additional 67-bp larger fragment that was confirmed with Sanger sequencing (Fig. S5C). Sanger sequencing of this genomic region confirmed a variant at the border of the aberrant splicing event, c.1569+339A>G (Table 1 and Fig. 2b). In silico analysis supported that this variant generated a new splicing acceptor site (Fig. S3).
In NM-U2, NM-U3, and NM-U4, two aberrant splicing events in NEB were common among the three samples: exonic splice gain leading to the 3′-side 5-bp shortening of exon 175 (Fig. 2c, left) and exon 175 skipping (Fig. 2c, right). We also found a 5-bp exon 131 extension caused by c.20158–6A>G in NM-U2 (see the legend of Table 1, S1, and Fig. S5A). Manual inspection with Integrative Genomic Viewer (IGV)31 of reads in the region of exon 175 identified additional potential aberrant splicing events in NM-U2, NM-U3, and NM-U4: increased out-of-frame intron 175 retention (Fig. 2c, left, arrow), in-frame exon 175 skipping (Fig. 2c, right; read count in arc with asterisk: 76, 3966, 2773, and 1372 in NP, NM-U2, NM-U3, and NM-U4, respectively), and in-frame skipping of exons 175 to 176 (Fig. 2c, right: read count: 66, 1061, 516, and 998 in NP, NM-U2, NM-U3, and NM-U4, respectively). Subtle but consistent aberrant splicing comprising exonic splice gain leading to 3′-sided out-of-frame 5-bp shortening of exon 175 and exon 176 skipping (Fig. 2c, right: read count: 48, 35, and 86 read counts in NM-U2, NM-U3, and NM-U4, respectively) was recognized. Furthermore, IGV inspection revealed that the reads from the intron 175 retention had a possible variant at the last nucleotide of exon 175, c.24684G>C (p.Ser8228Ser) in NM-U2, NM-U3, and NM-U4, while most reads of normal transcripts excising intron 175 did not possess this variant (Fig. 2d). Sanger sequencing confirmed the heterozygous variant (c.24684G>C) in NM-U2, NM-U3, and NM-U4. In silico analysis indicated that the scores of the wild-type splicing donor site were below the default thresholds, but those of the mutant allele were lower than those of the wild-type allele (Fig. S3). RT-PCR of cDNA from exons 173 to 177 followed by cloning, colony PCR, and Sanger sequencing confirmed an exonic splice gain leading to 3′-side 5-bp shortening of exon 175 and exon 175 skipping in NM-U2 (Fig. S4D). To confirm the increased intron 175 retention in the c.24684G>C allele, we performed RT-PCR with another set of primers aligning to exon 174 and the intron 175–exon 176 border. The RT-PCR amplicons contained the c.24684G>C allele more abundantly than the wild-type allele (Fig. S4E).
In NM-U6, the one candidate in NEB exhibited exon 54 skipping caused by the heterozygous c.7228–2A>G in NEB previously detected by ES in NM-U6 (Table 1, S1, and Fig. S5B). We could find no other splicing events that could be causative of NM in NM-U5 and NM-U6.
We analyzed overall NEB expression abundance in the RNA-seq data to evaluate the effects of the variants in NM-U1 to NM-U4 (Fig. 3a), together with the data of two NM-D with homozygous truncating variants: NM-D1, c.25234C>T (p.Arg8412*) and NM-D2, c.24786C>G (p.Tyr8262*); both were previously identified with ES. We found no significant decrease in total transcripts in the NM-U cases or in the positive control samples with biallelic truncating variants, except for NM-D2 and NM-U3, both of which had low z-scores (−2.72 and −1.95, respectively) (Fig. 3a). We also checked allelic imbalance in NEB expression at heterozygous variant sites. As a result, we found that allelic balance in the expression of the NEB variants in NM-U1 to NM-U4 deviated from that of variants of other genes of comparable read depth (Fig. 3b). The deviations were statistically significant in two or more NEB variants in NM-U1 to NM-U4 (Fig. 3b).

NEB expression analysis. (a) NEB expression from the RNA-seq data. Sample names are indicated below each bar. The z-scores are indicated above each bar. Z-scores were calculated based on the mean and standard deviations of the six undiagnosed myopathy of nonspecific muscle pathology (NP) cases. Y-axis: ratio of sense strand reads of NEB per total reads uniquely mapped to the genome. (b) Allelic imbalance of NEB transcripts in the RNA-seq data. The ratios of reads mapped to the altered allele versus the wild-type allele at heterozygous single-nucleotide variants (SNVs) were analyzed. Gray dots: SNV of all protein-coding genes except NEB; blue dots: SNV of NEB; red dots: the statistically significant SNV of NEB. X-axis: mean depth of altered and wild-type alleles. Y-axis: Log2 ratio of reads mapped to altered allele versus wild-type allele. (c) Position of NEB variants in cases for which the NEB expression was analyzed in (a). Variants in NM-U1 to NM-U4 and NM-D2 are at alternatively spliced exons. Horizontal bars: variant position, black bars: exons skipped in alternative transcripts. IF in-frame, OoF out-of-frame.
We checked whether the two variants identified by RNA-seq, c.1569+339A>G and c.24684G>C (p.Ser8228Ser), were registered in variant databases. c.1569+339A>G was not found in the disease-related variant databases nor in variant databases of normal populations containing data from genome sequencing (GS) (ToMMo or gnomAD). c.24684G>C was not found in disease-related variant databases but was identified in variant databases from the normal population. The minor allele frequencies (MAFs) were as follows: 0.00012 (2/16420) in East Asian population in gnomAD, 0.0028 (20/7104) in ToMMo, and 0.001 (2/2000) in HGVD.
Because of the relatively high frequency of c.24684G>C (p.Ser8228Ser) in Japanese controls of ToMMo, we further screened for this variant in ES data from Japanese NM cases. We identified the same variant in four cases: NM-U7, NM-U8, NM-U9, and NM-U10 and confirmed the variants by Sanger sequencing (Table 1). These patients each had an additional variant in NEB: c.19102–2A>G in NM-U7, c.23847+164A>G in NM-U8, c.7755del (p.Ser2585Argfs*34) in NM-U9, and c.9046C>T (p.Arg3016*) in NM-U10. Nishikawa et al. previously performed target sequencing for NM-U9 and NM-U10 and also reported these two variants (corresponding to cases 70 and 71 in their paper, respectively).26 All these variants, except for c.9046C>T (p.Arg3016*), were absent from the disease-related variant databases and the variant databases of normal populations. c.9046C>T (p.Arg3016*) had MAF of 0.000032 in gnomAD and reported as pathogenic in ClinVar and HGMD. In silico prediction tools suggested that 19102–2A>G resulted in the loss of the wild-type splice acceptor site, while c.23847+164A>G created a novel splicing donor site (Fig. S4F and S2G). RT-PCR confirmed that both variants resulted in aberrant splicing: in-frame exon 123 skipping (p.Val6368_Ser6402del) and intron 122 retention (p.Ile8560delinsArgLeuProValLeuLeuPheGlnGlyGlnThrIleSerCysLeuTyrValPheSerAsnAsnArgGlyGlu*) leading to a stop-gain in NM-U7 (Fig. S4F), and an out-of-frame exon 165 extension (p.Met7951Alafs*72) in NM-U8 (Fig. S4G).
Of the eight cases diagnosed in this study, seven cases were from a cohort of 197 NM cases that included 29 cases (14.7%) with pathogenic variants confirmed previously. Thus, this study successfully increased the diagnostic rate from 14.7% to 18.3% [=(7 + 29)/197].
DISCUSSION
In this study, we established an RNA-seq analytic workflow to efficiently detect aberrant splicing events and improve the diagnostic rate for muscle diseases, and we validated its usefulness in several unsolved NM cases. Previously, Kremer et al. and Cummings et al. described similar workflows for narrowing down candidate aberrant splicing events for the diagnosis of Mendelian diseases.10,11 Kremer’s workflow solely relied on p values of LeafCutter, and it set a strict threshold for the adjusted p value at<0.05, corresponding approximately to the nonadjusted p value of 0.0001 in our study. This strict threshold minimized the number of candidate aberrant splicing events (median: 5). However, because genes with lower expression levels tend to have larger p values, this strict threshold missed all of the aberrant splicing events in genes with lower expression: DMD and COL6A2 except for the one caused by c.1572+1G>C in COL6A2 (TPM median in the GTEx Portal: 25 in DMD and 58 in COL6A2). This strict threshold also missed two aberrant splicing events even in NEB (c.1569+339A>G [p.Leu524Phefs*9] in NM-U1, and c.20158–6A>G [p.Leu6721Argfs*11] in NM-U2), which is a highly expressed gene (TPM median in GTEx: 858) (Table S1). On the other hand, the workflow by Cummings et al. may be highly sensitive because it detected aberrant splicing events even in low-expression genes such as DMD and POMGNT1 (MIM 606822) (TPM median in GTEx: 29) (ref. 11). This workflow is, however, highly complicated, as it relies on many control samples from GTEx and information on several parameters as follows: (1) the number of samples in which the aberrant splicing event is present; (2) the number of reads supporting the aberrant splicing event; (3) the normalized value of the reads supporting the event, which is similar to ψ; and (4) the normalized value needs to be maximum in the sample and twice as much as the next highest sample. In addition, the number of aberrant splicing candidates produced by this workflow was 190 (median) for all genes beyond casual screening.11 Thus, the Cummings workflow is complicated and its specificity is not high. Our workflow essentially combines these two methods (Fig. 1d). (1) Our workflow may detect abnormal splicing events even in low-expression genes because we optimized the screening threshold comprising the ψ-D, ψ-FC, and p values using aberrant splicing events of the low-expression genes DMD and COL6A2 (Fig. 1d). (2) Our workflow is simple to apply because it uses only three parameters: ψ-D, ψ-FC, and the p value. (3) Our workflow requires only a small number of control samples (four NP cases in this study). (4) The number of aberrant splicing candidates produced by the workflow in our study was much lower than that described by Cummings et al. This could be attributed to an additional criterion, the p value of LeafCutter. However, the low number of candidates produced might also have been partly due to the threshold settings being too strict in our study (ψ-D: 0.1; ψ-FC: 20) because the number of positive controls (only eight aberrant splicing events) might have been insufficient (Fig. 1d). Overall, our workflow is easily applied but may have a better balance of sensitivity and specificity compared with the previous two workflows.
Nishikawa et al. previously reported a missense variant, c.20131C>T (p.Arg6711Trp), in NM-U3, NM-U9, and NM-U10 (cases 69, 70, and 71 in their paper, respectively).26 We also identified c.20131C>T in NM-U2, NM-U4, NM-U7, and NM-U8. Because all these cases have both c.24684G>C (p.Ser8228Ser) and c.20131C>T (p.Arg6711Trp), these variants are most likely in the same allele. c.20131C>T (p.Arg6711Trp) has been reported as a variant of unknown significance.25,26 Our study suggested that c.24684G>C (p.Ser8228Ser) is most likely causative rather than c.20131C>T, because the variant causing aberrant splicing leads to an premature termination (p.Ser8228Valfs*17, p.Tyr8231Serfs*2, and p.Ser8228Ilefs*22), a popular pathomechanism of NEB pathogenic variants in NM (Table 1).
Using RNA-seq, we identified novel NEB pathogenic variants that had been missed by ES analysis. One of these pathogenic variants, c.24684G>C, had the highest MAF (0.0028) in the general Japanese population (Table 2). Note that c.20131C>T (p.Arg6711Trp), a variant of higher MAF, is unlikely to be pathogenic (see the above discussion). Indeed, we identified c.24684G>C (p.Ser8228Ser) in seven NM cases. A previous study also identified a deep-intronic pathogenic variant of COL6A2 with high MAF by RNA-seq, and the pathogenic variant explained 20% of undiagnosed cases of UCMD.11 Thus, RNA-seq can undoubtedly contribute toward solving a significant number of previously unsolved cases of muscle diseases.
We analyzed the effect of NEB pathogenic variants on its expression. We found no significant change in total NEB expression in any of the samples except for NM-D2 and NM-U3 (z-score: −2.72 and −1.95, respectively). This is partly explained by aberrant splicing leading to in-frame changes and/or mutated exons present only in alternative transcripts (Fig. 3c). Another reason may be the incomplete normalization of NEB expression. We normalized the number of reads mapped to NEB based on the total number of uniquely mapped reads to the genome. This ratio may be influenced by muscle condition at the biopsy site such as muscle degeneration and different cell/tissue infiltration. To address this potential problem, another normalization method will be required. We also analyzed the allelic balance of NEB expression (Fig. 3b). Allelic imbalance was recognized for NEB expression in all analyzed samples, NM-U1 to NM-U4. This result suggested that the expression of either or both alleles was dysregulated, consistent with the presence of biallelic truncating variants in NM-U1 to NM-U4.
In summary, we established a new RNA-seq analytic workflow for narrowing down candidate aberrant splicing events to a tractable number for further assessment. Using this workflow, we could identify the most frequent but unrecognized NEB pathogenic variant and successfully demonstrated that RNA-seq has significant potential for resolving many undiagnosed Mendelian disease cases.
References
Trujillano D, Bertoli-Avella AM, Kumar Kandaswamy K, et al. Clinical exome sequencing: results from 2819 samples reflecting 1000 families. Eur J Hum Genet. 2017;25:176–182.
Monies D, Abouelhoda M, AlSayed M, et al. The landscape of genetic diseases in Saudi Arabia based on the first 1000 diagnostic panels and exomes. Hum Genet. 2017;136:921–939.
Rabbani B, Tekin M, Mahdieh N. The promise of whole-exome sequencing in medical genetics. J Hum Genet. 2014;59:5–15.
Srivastava S, Cohen JS, Vernon H, et al. Clinical whole exome sequencing in child neurology practice. Ann Neurol. 2014;76:473–483.
Miyatake S, Matsumoto N. Genetics: clinical exome sequencing in neurology practice. Nat Rev Neurol. 2014;10:676–678.
Biesecker LG, Green RC. Diagnostic clinical genome and exome sequencing. N Engl J Med. 2014;370:2418–2425.
Naruto T, Okamoto N, Masuda K, et al. Deep intronic GPR143 mutation in a Japanese family with ocular albinism. Sci Rep. 2015;5:11334.
Nozu K, Iijima K, Nozu Y, et al. A deep intronic mutation in the SLC12A3 gene leads to Gitelman syndrome. Pediatr Res. 2009;66:590–593.
Dehainault C, Michaux D, Pages-Berhouet S, et al. A deep intronic mutation in the RB1 gene leads to intronic sequence exonisation. Eur J Hum Genet. 2007;15:473–477.
Kremer LS, Bader DM, Mertes C, et al. Genetic diagnosis of Mendelian disorders via RNA sequencing. Nat Commun. 2017;8:15824.
Cummings BB, Marshall JL, Tukiainen T, et al. Improving genetic diagnosis in Mendelian disease with transcriptome sequencing. Sci Transl Med. 2017;9. Pii eaal5209, https://doi.org/10.1126/scitranslmed.aal5209. Accessed 21 November 2018.
Dobin A, Davis CA, Schlesinger F, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21.
Katz Y, Wang ET, Airoldi EM, Burge CB. Analysis and design of RNA sequencing experiments for identifying isoform regulation. Nat Methods. 2010;7:1009–1015.
Ihaka R and Gentleman R. R: a language for data analysis and graphics. J. Comp. Graph. Stat. 1996;5:299–314.
Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550.
Pervouchine DD, Knowles DG, Guigo R. Intron-centric estimation of alternative splicing from RNA-seq data. Bioinformatics. 2013;29:273–274.
Li YI, Knowles DA, Humphrey J, et al. Annotation-free quantification of RNA splicing using LeafCutter. Nat Genet. 2018;50:151–158.
Fujita A, Isidor B, Piloquet H, et al. De novo MEIS2 mutation causes syndromic developmental delay with persistent gastro-esophageal reflux. J Hum Genet. 2016;61:835–838.
Hoffman EP, Brown RH Jr, Kunkel LM. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell. 1987;51:919–928.
Higuchi I, Shiraishi T, Hashiguchi T, et al. Frameshift mutation in the collagen VI gene causes Ullrich’s disease. Ann Neurol. 2001;50:261–265.
Pelin K, Hilpela P, Donner K, et al. Mutations in the nebulin gene associated with autosomal recessive nemaline myopathy. Proc Natl Acad Sci U S A. 1999;96:2305–2310.
Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–291.
Higasa K, Miyake N, Yoshimura J, et al. Human genetic variation database, a reference database of genetic variations in the Japanese population. J Hum Genet. 2016;61:547–553.
Nagasaki M, Yasuda J, Katsuoka F, et al. Rare variant discovery by deep whole-genome sequencing of 1,070 Japanese individuals. Nat Commun. 2015;6:8018.
Tsunoda K, Yamashita T, Motokura E, et al. A patient with slowly progressive adult-onset nemaline myopathy and novel compound heterozygous mutations in the nebulin gene. J Neurol Sci. 2017;373:254–257.
Nishikawa A, Mitsuhashi S, Miyata N, Nishino I. Targeted massively parallel sequencing and histological assessment of skeletal muscles for the molecular diagnosis of inherited muscle disorders. J Med Genet. 2017;54:104–110.
Cartegni L, Wang J, Zhu Z, Zhang MQ, Krainer AR. ESEfinder: a web resource to identify exonic splicing enhancers. Nucleic Acids Res. 2003;31:3568–3571.
Reese MG, Eeckman FH, Kulp D, Haussler D. Improved splice site detection in Genie. J Comput Biol. 1997;4:311–323.
Hebsgaard SM, Korning PG, Tolstrup N, Engelbrecht J, Rouze P, Brunak S. Splice site prediction in Arabidopsis thaliana pre-mRNA by combining local and global sequence information. Nucleic Acids Res. 1996;24:3439–3452.
Lehtokari VL, Kiiski K, Sandaradura SA, et al. Mutation update: the spectra of nebulin variants and associated myopathies. Hum Mutat. 2014;35:1418–1426.
Thorvaldsdottir H, Robinson JT, Mesirov JP. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform. 2013;14:178–192.
Acknowledgements
See supplementary information.
Author contributions:
K.H.: literature review, data collection, and drafting the manuscript; E.K., Y.T., S.Mit., K.I., A.N.A., A.F., E.I., Y.U., N.T., Y.A., Y.M., M.O., M.N., T.M., N.Mi., H.S., and A.I.: data collection and manuscript revision; S.Miy., A.T., I.N., and N.M.: supervision of all aspects, including study design, data interpretation, and manuscript preparation.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Disclosure
The authors declare no conflicts of interest.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Hamanaka, K., Miyatake, S., Koshimizu, E. et al. RNA sequencing solved the most common but unrecognized NEB pathogenic variant in Japanese nemaline myopathy. Genet Med 21, 1629–1638 (2019). https://doi.org/10.1038/s41436-018-0360-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-018-0360-6
Keywords
This article is cited by
-
Reprograming skin fibroblasts into Sertoli cells: a patient-specific tool to understand effects of genetic variants on gonadal development
Biology of Sex Differences (2024)
-
Molecular signatures of inherited and acquired sporadic late onset nemaline myopathies
Acta Neuropathologica Communications (2023)
-
Nemaline myopathy: reclassification of previously reported variants according to ACMG guidelines, and report of novel genetic variants
European Journal of Human Genetics (2023)
-
Genome sequencing and RNA sequencing of urinary cells reveal an intronic FBN1 variant causing aberrant splicing
Journal of Human Genetics (2022)
-
Feasibility of predicting allele specific expression from DNA sequencing using machine learning
Scientific Reports (2021)
