
Abstract
Purpose
Vascular Ehlers–Danlos syndrome (vEDS) is a rare inherited autosomal dominant disorder caused by COL3A1 pathogenic variants. A high percentage of de novo cases has been suggested. Part of it could be due to parental mosaicism, but its frequency is unknown.
Methods
This retrospective study included a large series of COL3A1-confirmed vEDS probands with family information. The frequency of de novo cases was evaluated and the distribution of the type of variants was compared according to the mode of inheritance. The COL3A1 mosaicism was studied by deep targeted next- generation sequencing (NGS) from parental blood DNA.
Results
Out of 177 vEDS probands, 90 had a negative family history, suggesting a high rate (50.8%) of de novo pathogenic variants, enriched in the more severe COL3A1 variants (no null variant). Among those, both parental DNA were available in 36 cases and one parental DNA in 18 cases. NGS detected only one mosaicism from maternal blood DNA (allelic ratio 18%), which was confirmed in saliva (allelic ratio 22%).
Conclusion
vEDS is characterized by a high frequency of de novo pathogenic variants. Parental mosaicism is rare (2–3%), but should be systematically searched with targeted NGS, taking into account its importance in genetic counseling.
Similar content being viewed by others

INTRODUCTION
Vascular Ehlers–Danlos syndrome (vEDS, OMIM 130050) is a rare (prevalence estimated at 1/150,000) inherited autosomal dominant (AD) disorder that results from pathogenic variants at the COL3A1 gene (OMIM 120180), which encodes the proα1(III) chain of type III collagen.1 Minor signs such as easy bruising, translucent skin with visible veins, and characteristic facial features are often present in childhood. Patients are exposed to life-threatening complications starting in young adulthood with a mean age of 30 years at the first major complication, depending on the type of COL3A1 pathogenic variants.2,3 Most often, these major symptomatic events correspond to spontaneous arterial dissections and ruptures, bowel perforations, and less frequently to pneumothorax and uterine ruptures.3 Up to now, no consequence of COL3A1 pathogenic variants on fertility has been suggested, but an increased risk of death exists in affected women in the peripartum period,4 when compared with the general population. The severity of vEDS could thus lead to a lower reproductive fitness. Finally, even though the disease expressivity is variable, the penetrance is complete,5 making the family history an interesting tool to suggest a parental transmission.
To date, more than 600 unique pathogenic variants have been identified in the COL3A1 gene as causing vEDS.6 Most of them are private, spread along the coding part of the collagen III triple helix. These include glycine substitutions within the triple helical region of type III collagen, splice-site, frameshift, nonsense, or large deletion variants. vEDS is inherited in an AD manner and exceptional cases of biallelic inheritance have been reported.7,8 In one US series, the frequency of probands with a negative family history was 50%, suggesting a high rate of de novo pathogenic variants.2 However, no molecular parental study was performed to confirm this estimation. Large human genome studies and analyses of parent–offspring trios have allowed an estimation of approximately 50–100 de novo molecular variants per generation and individual.9,10 A recent study performed in a large sample of patients with various inherited diseases that included parental samples showed a high percentage (68%) of de novo pathogenic variants in AD diseases.11 De novo pathogenic variants are well known to play an important role in severe early-onset diseases because of their impact on the reproductive fitness. Thus their role in late-onset disorders like vEDS (adult onset versus pediatric onset) is smaller.9
The use of next-generation sequencing (NGS) offers the possibility to detect more precisely the presence of parental mosaicism.12 Recent genome sequencing studies allowed to evaluate the extent of mosaicism for de novo variants. A trio-based study showed that 6.5% of de novo pathogenic variants were actually postzygotic mosaic in probands with intellectual disability, and that 0.1% of de novo pathogenic variants resulted from parental mosaicism.13 More recently, the analysis of data in three families showed that 4% of apparently de novo variants originate from parental gonosomal mosaicism detectable in blood.14 In vEDS, the frequency of parental somatic mosaicism has been estimated at 15% without molecular study supporting this estimation.15 Since the identification of COL3A1, only five cases of COL3A1 somatic and germline mosaicism have been reported in early and/or partially documented publications.16,17,18,19,20 All these cases were single observations and asymptomatic relatives of probands. To the best of our knowledge, to date, no systematic study has been performed to determine the true frequency of COL3A1 mosaicism.
We assessed the frequency of de novo COL3A1 pathogenic variants in a French series of 177 vEDS families and confirmed the previous estimation of this frequency. In 54 families with a de novo pathogenic variant, we screened COL3A1 somatic mosaic by deep targeted NGS. The results suggested mosaicism is a rare event in vEDS.
MATERIALS AND METHODS
Study population
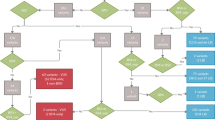
To determine the frequency of de novo pathogenic variants, we included all vEDS probands referred between January 2000 and June 2017 to the Genetics Department of the Georges Pompidou European Hospital (Paris, France) for molecular analysis and harboring a COL3A1 pathogenic variant. Family history (FH) was assessed through questionnaire and/or direct examination and genetic testing in the parents/relatives. Positive FH was considered as confirmed if (1) the mother or the father was heterozygous for the COL3A1 pathogenic variant, or (2) the recurrence in the siblings was molecularly proven with a parental transmission in the questionnaire, or (3) the recurrence in the siblings was molecularly proven without a parental transmission in the questionnaire. Positive FH was considered as apparent (i.e., without molecular confirmation) in three conditions: (1) only one parental DNA was available and negative for the COL3A1 pathogenic variant that seemed to be transmitted by the other parent in the questionnaire, or (2) FH was assessed in the questionnaire and not confirmed with one/several relatives negative for the COL3A1 pathogenic variant, or (3) FH was assessed only in the questionnaire with no family screening (Fig. 1 and Supplemental Fig. 1). Sporadic status was considered as confirmed only when both parental DNA were available and were not heterozygous for the COL3A1 pathogenic variant. Sporadic status was considered as apparent in three conditions when no FH was mentioned in the questionnaire: (1) one parental DNA was available and not heterozygous for the COL3A1 pathogenic variant, or (2) no parental DNA was available and one/several relatives were negative for the COL3A1 pathogenic variant, or (3) neither parental DNA nor DNA of relatives were available (Fig. 1).

Flowchart of the study. *See Supplemental Fig. 1 for further details. NGS next-generation sequencing.
When the pathogenic variant identified in the proband was not found at heterozygous stage on the DNA of parents with Sanger sequencing, the parental DNA was included in NGS analysis to detect and estimate the frequency of mosaicism. Probands were also included as positive control of the family pathogenic variant for this analysis.
The frequencies of de novo pathogenic variants and mosaicism were calculated in two different ways to take into account first only confirmed inheritance status and second both confirmed and apparent inheritance status.
The patients’ clinical characteristics were staged as major or minor diagnostic criteria according to the 2017 New York criteria.1 The study was reviewed and approved by Assistance Publique – Hôpitaux de Paris. Written informed consent for the genetic study was obtained, and genetic testing was performed in accordance with French legislation regarding genetics diagnostics tests (French bioethics law 2004-800).
Genetic analysis
Genomic DNA was isolated from leukocyte pellets with Qiamp DNA Blood Midi Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions; and from Oragene samples with ethanol precipitation protocol and prepIT L2P reagent (DNA Genotek, Ottawa, Canada).
Sanger sequencing: COL3A1 exons were amplified by polymerase chain reaction (PCR) with specific primers (Supplemental Table 1) and then sequenced using BigDye Terminator kit v3.1 cycle sequencing kits and run on an ABI Prism 3730XL DNA Analyzer (Perkin Elmer Applied Biosystems, Foster City, CA). DNA variants were identified using Sequencher software.
NGS sequencing: COL3A1 exons enrichment was performed by PCR with the same specific primers. Then PCR products were cleaned up in a two-step protocol: first using a combination of Exonuclease I and FastAP™ Thermosensitive Alkaline Phosphatase (Thermo Fisher Scientific, Waltham, MA), second using a 0.8X Agencourt AMPure XP bead cleanup (Beckman Coulter, Indianapolis, IN) to eliminate PCR primers as much as possible. Purified PCR products were quantified using Qubit dsDNA BR Assay Kit (Thermo Fisher Scientific, Waltham, MA). Next, libraries were prepared with Nextera XT DNA Library Prep Kit (Illumina, San Diego, CA) according to the manufacturer’s instructions and analyzed on MiSeq (Illumina, San Diego, CA).
The 144 samples were pooled and loaded on the same flow cell (V2-500 cycles) using Nextera XT index kit V2 sets A and B (Illumina, San Diego, CA) to have enough combinations.
NGS analysis: Bioinformatic analysis was performed using an in-house pipeline developed from open source tools. After demultiplexing, sequences were aligned to the reference human genome hg19 using the Burrows–Wheeler Aligner (BWA). Downstream processing was carried out with the Genome Analysis Toolkit (GATK), SAMtools, and Picard, following documented best practices (http://www.broadinstitute.org/gatk/guide/topic?name=best-practices). Variant calls were made with the GATK Unified Genotyper. The annotation process was based on the latest release of the Ensembl database. Variants were annotated and analyzed using the Polyweb software interface designed by the Bioinformatics platform of University Paris Descartes.
Statistical analysis
We compared the types of variants between the patients with an inherited pathogenic variant and the patients with a de novo pathogenic variant using Chi-squared or Fisher's exact test (when count was insufficient in the contingency table). A p value ≤0.05 was considered significant.
Pathogenic variants were classified in three groups according to previous genotype–phenotype correlation:3glycine substitution within the triple helix (group I); splice-site variants, deletions, duplications, indels, all in frame (group II); and variants leading to haploinsufficiency (group III). The comparison was made first for confirmed inheritance status, and second for confirmed and apparent inheritance status.
RESULTS
Estimation of de novo pathogenic variants frequency
From January 2000 to June 2017, 194 vEDS probands were diagnosed with an identified COL3A1 pathogenic variant. Seventeen cases with no available family information were excluded thus 177 probands were included in this study (Fig. 1). Among them, 87 (n = 87/177 = 49.2%) had a FH of which 49 (n = 49/87 = 56.3%) were confirmed at molecular level (identification of the COL3A1 pathogenic variant in parents and/or relatives, details about family screening, and family information from the questionnaire are available in Supplemental Fig. 1). In the remaining 90 cases, both parental DNA were tested in 36 cases (n = 36/90 = 40.0%), which were shown to have a de novo pathogenic variant (Fig. 1). Thus, in our series the frequency of confirmed de novo pathogenic variants, where both parents confirmed no variant, was estimated at 42.4% (n = 36/36 + 49 = 42.4%) and the frequency of confirmed and apparent de novo pathogenic variants was estimated at 50.8% (n = 90/90 + 87 = 50.8%, Table 1).
Inheritance pattern and distribution of the type of variant
In the 177 families with family information, 103 (58.2%) glycine substitutions, 58 (32.8%) splice-site variants or deletions or duplications or indels, 15 (8.5%) variants leading to haploinsufficiency, and 1 variation of unknown significance (VUS) were identified (Fig. 2 and Supplemental Table 2). The majority of them (n = 130/177 = 73.5%) were private. The unique case with a VUS was excluded from this comparison. The type of variant was associated with its de novo or inherited character for confirmed genetic status (p = 2.1e−5) and for both apparent and confirmed genetic status (p = 2.7e-4, Table 2).

Types of variants of the 177 families included and their position along the COL3A1 gene. Boxes represent exons of the COL3A1 gene (NG_007404.1). The color code indicates the type of variants (glycine substitutions in black, splice-site variants in red, deletions and indels in green, variants leading to haploinsufficiency in blue, and variants of uknown significance [VUS] in yellow). The complete nomenclature of each variant can be found in Supplemental Table 2. The numbers in bold indicate the number of times the variants were found.
Impact of vEDS on the reproductive fitness
We evaluated the reproductive fitness in vEDS by studying the relative number of children born to affected and unaffected cases in the same families. The comparison was made distinguishing male and female. We had the information for 119 affected vEDS women who had an average number of children of 1.4, whereas 97 unaffected vEDS women belonging to the same families had an average number of children of 2.3 (Student's test, p = 1.1e-9).
Likewise, we had the information for 97 affected vEDS men who had an average number of children of 1.1, whereas 86 unaffected vEDS men had an average number of children of 1.9. Thus the average number of children was also significantly lower for affected men than for unaffected men (Student's test, p = 5.7e-6).
Then we determined the average number of children in families after a first affected or unaffected child. Seventy-five families had a first affected child with an average number of further children of 1.17. The information was available for only 28 families after a first unaffected child revealing the lack of information for unaffected siblings. The average number of children after the first unaffected child was 1.14. Thus the average number of further children was not significantly different for affected or unaffected first child (Student's test, p = 0.85).
Estimation of COL3A1 mosaicism with NGS analysis
In the 90 apparent sporadic cases, the DNA of both parents was available for 36 families forming trios with the proband and the DNA of one parent was available for 18 families forming duos with the proband (13 mothers and 5 fathers, Fig. 1) with the restriction of the use of blood DNA.
Ninety [90 = (36 × 2) + (18 × 1)] parents were included in the NGS sequencing analysis to estimate the frequency of parental somatic mosaicism (Fig. 1 and Supplemental Table 3).
One library failed, corresponding to the positive control of family 22. The mean sequence coverage per base was approximately 60,000X with an extremely high heterogeneity (range: 443–375,925) in the 143 libraries (Supplemental Fig. 2 and Supplemental Table 4).
One mosaic was identified in a mother. The NGS analysis showed the presence of the pathogenic variant c.1194+2T>A with an allele ratio of 18% in the leukocytes DNA and 22% in the saliva (Oragene sample, Fig. 3b, d). The blood samples from both parents were also screened by direct Sanger sequencing for the COL3A1 pathogenic variant. A small peak was observed in the chromatogram of the mother’s leukocytes DNA (Fig. 3c). The pathogenic variant was not detected in the father.

Detection of one COL3A1 mosaicism in a mother of vascular Ehlers–Danlos syndrome (vEDS) proband. (a) Pedigree of the family. Circles indicate females, squares indicate males. The proband (II-1) is indicated with a black arrow. Current age is indicated above each individual (y = years). (b) Reads of deep targeted next-generation sequencing (NGS) with the COL3A1 mosaic. (c) Sanger sequencing showing the causal pathogenic variant. Electrophoregrams of COL3A1 intron 17 obtained from the proband (II-1) and the mother (I-2) blood leukocytes genomic DNA (gDNA). (d) Allelic ratio of the mosaic in blood and saliva.
Thus, the frequency of parental COL3A1 somatic mosaicism was estimated at 1.9% [n = 1/(36 + 18) = 1.9%] if one considers the 54 families with an apparent de novo pathogenic variant. Another calculation method was used with an estimated frequency of 2.8% (n = 1/36) if one considers only the 36 families with both available parental DNA and a confirmed de novo pathogenic variant (Table 1).
Among the 87 family cases, recurrence in siblings was confirmed in 1 family and apparent in 4 families. This recurrence was supposed to be due to dominant parental transmission but could also be due to parental mosaicism. Unfortunately this unlikely hypothesis could not be tested because parental DNA was not available for these families. If we supposed these five occurrences were due to parental mosaicism, and then were included to estimate the parental mosaicism, the frequency of parental somatic mosaicism could be estimated at 14.6% [n = (1 + 5)/(36 + 5)].
Mosaic case description
The mosaic was found in a 68-year-old woman whose daughter was diagnosed with vEDS at the age of 39 in the presence of a dissecting aneurysm of the left external iliac artery, a spontaneous rupture of the gallbladder, thin fragile skin, easy bruising, papyraceous scars, and evocative facial morphotype. The diagnosis was molecularly confirmed with the identification of a heterozygous pathogenic variant c.1194+2T>A, p.(?) in COL3A1 intron 17 leading to exon 17 skipping.
A parental mosaicism or a family pathogenic variant should have been suspected because of the family. The proband had a young brother who died after hemorrhage due to road traffic injury. This brother presented several minor signs: a characteristic facial appearance, a translucent and thin skin, hypermobility of joints with several sprains, and a premature delivery. This raised recurrence in this family (Fig. 3a). Furthermore, the mother had several minor criteria suggestive of the disease: thin and translucent skin, acrogeria, easy bruising, characteristic facial appearance, hypermobility of small joints with dislocations of the thumbs, and gingival recession, but no major complication.
DISCUSSION
This study corroborates that the proportion of affected individuals with a de novo pathogenic variant is high, with an estimated frequency of 42.4–50.8%. In a previous estimation,2 FH was confirmed with family screening or estimated through questionnaire and/or relatives' clinical evaluation. However, the exact numbers of cases with confirmed and apparent inheritance status were absent in this study. Lack of family screening and information highlights the difficulties to determine the correct frequency of de novo COL3A1 pathogenic variants and explains the variation in our estimates. However, no strong difference was observed between the two estimates (42.4% vs. 50.8%) assuming no strong bias in the rough estimation of 50% for de novo transmission. The collection of parental samples to search for de novo pathogenic variants is a major challenge in adult-onset disorders.21 In addition a family bias of recruitment is possible because vEDS is a rare disease poorly understood by the majority of physicians and sporadic cases could be missed because signs and symptoms of vEDS are not well known and particularly in absence of family history suggestive of genetic disorder. However, our results are coherent with those observed in a large sample of patients with various inherited AD diseases.11
The relationship between the fitness effect of a pathogenic variant and the proportion of de novo pathogenic variants observed in a dominantly inherited disorder is well illustrated in vEDS. The frequency of de novo pathogenic variants in a particular disease is mainly determined by the size of the mutational target (i.e., the coding sequence of a gene) for the disease.10 In the case of a monogenic disorder such as vEDS, the probability of a new mutational event at the COL3A1 gene is very low (size of COL3A1 complementary DNA [cDNA]/size of genomic coding sequence = 1.4e-4) explaining why this disorder is rare in the general population (prevalence estimated at 1/150,000). Three other factors can influence de novo pathogenic variants' frequency: (1) genetic homogeneity, (2) dominant inheritance, and (3) marked negative fitness effect of pathogenic variants.10 The stronger these factors are, the more they reduce the impact of inherited factors.10 These three factors might be at play in vEDS because COL3A1 is the only causal gene, the transmission is dominant, and the life-threatening complications occur at a mean age of 30 years.2,3 Furthermore, considering AD monogenic disorders, the frequency of de novo pathogenic variants depends mainly on the fitness effect of pathogenic variants. This frequency can reach 100% for severe pediatric disorders22 and decreases with the age onset and the severity of the disorder. In that regard, the significant bias (p = 2.1e-5 and 2.7e-4) observed in the distribution of the type of pathogenic variants in family cases compared with sporadic cases can be explained, because the variants leading to haploinsufficiency and to a milder phenotype3 were not observed in the de novo confirmed cases. To our knowledge, such a significant bias has never been described before for another monogenic AD disorder.
The reproductive fitness in vEDS was evaluated by studying the relative number of children born to affected and unaffected cases in the same families. The comparison was made distinguishing male and female because women have the added issue of pregnancy-related complications. The average number of children was significantly lower for both affected men and women than for unaffected men and women. This difference could imply that vEDS reduces the reproductive fitness. However, the interpretation needs to be cautious: if an effect of the pathology on the reproductive fitness could be suggested, the lower number of children for affected cases could also indicate a choice of these patients to have no or fewer children taking into account a willingness to not transmit the disease. But the average number of further children after a first affected or unaffected child was not significantly different, which suggests a biological effect on reproductive fitness is more likely than societal restraints to not transmit the disease.
As increasing paternal age is known to positively correlate with the risk of de novo pathogenic variant because of germinal mosaicism,23 we also studied the effects of paternal age. The median paternal age was estimated at 28 for family cases and at 29.5 for sporadic cases with no significant difference. So we failed to highlight an effect of paternal age in our cohort; however, one limiting factor is sample size.
Our study is the first one to evaluate accurately the frequency of COL3A1 parental somatic mosaicism in a large group of vEDS patients. Until now, the lack of sensitivity of direct Sanger sequencing for detecting a low disease/wild-type allelic ratio,24 and the rarity of the disease made difficult a precise estimation of COL3A1 mosaicism. Pepin et al. estimated COL3A1 parental somatic mosaicism at 15% 15 but no information was provided concerning the design of this study which has not been subjected to publication. Another study reported the detection of pathogenic mosaic variants in human genetic disorders with different inheritance patterns. Eight probands harboring a COL3A1 pathogenic variant were recruited. Among them only one was an apparent de novo case and was included with parents for COL3A1 NGS sequencing to detect mosaicism. This limited study suggested a high frequency of parental somatic mosaicism (n = 1/8 = 12.5%) 20. Our estimate is much lower (1.9–2.8%) and more consistent with the very small number of published COL3A1 mosaicism cases (n = 5). Our estimate is also consistent with the frequencies of parental somatic mosaicism determined in two genome sequencing studies;13,14 0.1% and 4% of parental somatic mosaicisms were found among de novo variants. Furthermore, the frequency of mosaicism seems to vary among different AD disorders and the higher the prevalence of de novo cases is, the higher the frequency of mosaicism should be. For example, the frequency of mosaic is approximatily 8% in NF1 25 and Alagille syndrome due to JAG1 pathogenic variants,26 while only few cases of mosaic were reported in Marfan syndrome.27 However, the number of parental mosaicisms could be underestimated due to five cases of recurrence in siblings that could not be molecularly investigated in our cohort. If these five cases were taken into account to estimate the frequency of parental somatic mosaicism, this could reach nearly 15%. But this hypothesis is very unlikely and could not be supported by the nonaffected status of the parents. Interestingly, the published COL3A1 mosaicism cases as well as ours were identified through a familial genetic screening secondary to the diagnosis made in a symptomatic proband. Two cases were asymptomatic,16,17 one case was not specified,20 and two cases had minor signs: one father shared keloid scars with his daughter, who presented a typical vEDS phenotype,18 and one maternal grandmother had a slight ectasia of thoracic aorta at 79 19). Our case had several minor criteria: thin and translucent skin, acrogeria, easy bruising, characteristic facial appearance, hypermobility of small joints with dislocations of the thumbs, and gingival recession, a combination of unspecific signs suggestive of vEDS only in the presence of a familial symptomatic case. To detect mosaicism, we used NGS, which is the most powerful and sensitive approach.28 A well-known shortcoming of NGS is the high rate of sequencing errors, which cannot be disregarded when identifying low-level somatic mosaicism. For example, Izawa et al.29 were able to detect 1% somatic mosaicism in the NLRP3 gene with 99.9% confidence when more than 350 reads were accumulated for each strand. We chose to use deep targeted NGS (average number of reads = 60,000X) to enhance sensitivity and accuracy of the detection of a given variant at the COL3A1 gene. However, the use of the Nextera XT technology led to a large heterogeneity of coverage (Supplemental Fig. 2). Despite this heterogeneity, only two samples of our series (mothers of families 2 and 25, Supplemental Table 4) did not reach the threshold of 2 × 350X at the pathogenic variant position. Because the wild-type allele was 100% at the pathogenic variant position for all parents’ DNA, we can assume that no mosaic was missed in our series (Supplemental Table 4).
One limitation of our retrospective study is that only peripheral DNA was available for all patients tested, thus limiting the sensitivity of the analysis. The availability of different somatic tissues (blood, saliva, dermal fibroblasts, and oral mucosa) would have helped to obtain a precise quantification of the mosaic level. It could even provide precisions for transmission risk to offspring in genetic counseling, particularly when the germinal cells are not available (e.g., in females). Finally, the availability of different tissues could also help to determine if a correlation exists between the extent of the mosaic and the severity of the phenotype. In the case of vEDS, the availability of dermal fibroblasts could help to determine if skin phenotype correlates to the presence/level of a cutaneous mosaic.
In conclusion, we determined the frequency of de novo COL3A1 pathogenic variants at 50% in a French series of 177 vEDS patients, confirming the high rate of de novo pathogenic variants in this rare and life-threatening condition. We further screened COL3A1 somatic mosaicism by deep targeted NGS in 54 families and identified only one COL3A1 case of leukocytes mosaicism in a mother with several diagnostic minor criteria but without spontaneous severe complication. We thus estimated the frequency of COL3A1 somatic mosaicism at 2–3% suggesting this mechanism is rare in vEDS. Despite this low frequency a systematic deep targeted NGS in parents of a vEDS de novo patient should be performed in the presence of minor signs taking into account its importance for accurate genetic counseling.
References
Malfait F, Francomano C, Byers P, et al. The 2017 international classification of the Ehlers-Danlos syndromes. Am J Med Genet C Semin Med Genet. 2017;175:8–26.
Pepin MG, Schwarze U, Rice KM, Liu M, Leistritz D, Byers PH. Survival is affected by mutation type and molecular mechanism in vascular Ehlers-Danlos syndrome (EDS type IV). Genet Med. 2014;16:881–888.
Frank M, Albuisson J, Ranque B, et al. The type of variants at the COL3A1 gene associates with the phenotype and severity of vascular Ehlers-Danlos syndrome. Eur J Hum Genet. 2015;23:1657–1664.
Pepin M, Schwarze U, Superti-Furga A, Byers PH. Clinical and genetic features of Ehlers-Danlos syndrome type IV, the vascular type. N Engl J Med. 2000;342:673–680.
Byers PH, Belmont J, Black J, et al. Diagnosis, natural history, and management in vascular Ehlers-Danlos syndrome. Am J Med Genet C Semin Med Genet. 2017;175:40–47.
Search unique variants - Ehlers Danlos Syndrome Variant Database - Leiden Open Variation Database. University of Leicester, UK: Raymond Dalgleish; 2008. https://eds.gene.le.ac.uk/home.php?select_db=COL3A1. Accessed March 1, 2018.
Plancke A, Holder-Espinasse M, Rigau V, Manouvrier S, Claustres M, Khau Van Kien P. Homozygosity for a null allele of COL3A1 results in recessive Ehlers-Danlos syndrome. Eur J Hum Genet. 2009;17:1411–1416.
Jørgensen A, Fagerheim T, Rand-Hendriksen S, et al. Vascular Ehlers-Danlos Syndrome in siblings with biallelic COL3A1 sequence variants and marked clinical variability in the extended family. Eur J Hum Genet. 2015;23:796–802.
Acuna-Hidalgo R, Veltman JA, Hoischen A. New insights into the generation and role of de novo mutations in health and disease. Genome Biol. 2016;17:241.
Veltman JA, Brunner HG. De novo mutations in human genetic disease. Nat Rev Genet. 2012;13:565–575.
Posey JE, Harel T, Liu P, et al. Resolution of disease phenotypes resulting from multilocus genomic variation. N Engl J Med. 2017;376:21–31.
Lupski JR. Genetics. Genome mosaicism—one human, multiple genomes. Science. 2013;341:358–359.
Acuna-Hidalgo R, Bo T, Kwint MP, et al. Post-zygotic point mutations are an underrecognized source of de novo genomic variation. Am J Hum Genet. 2015;97:67–74.
Rahbari R, Wuster A, Lindsay SJ, et al. Timing, rates and spectra of human germline mutation. Nat Genet. 2016;48:126–133.
Pepin MG, Murray ML, Byers PH. Vascular Ehlers-Danlos syndrome. In: Adam MP, Ardinger HH, Pagon RA, et al., eds. GeneReviews. Seattle (WA): University of Washington, Seattle; 1993.
Kontusaari S, Tromp G, Kuivaniemi H, Stolle C, Pope FM, Prockop DJ. Substitution of aspartate for glycine 1018 in the type III procollagen (COL3A1) gene causes type IV Ehlers-Danlos syndrome: the mutated allele is present in most blood leukocytes of the asymptomatic and mosaic mother. Am J Hum Genet. 1992;51:497–507.
Richards AJ, Ward PN, Narcisi P, Nicholls AC, Lloyd JC, Pope FM. A single base mutation in the gene for type III collagen (COL3A1) converts glycine 847 to glutamic acid in a family with Ehlers-Danlos syndrome type IV. An unaffected family member is mosaic for the mutation. Hum Genet. 1992;89:414–418.
Milewicz DM, Witz AM, Smith AC, Manchester DK, Waldstein G, Byers PH. Parental somatic and germ-line mosaicism for a multiexon deletion with unusual endpoints in a type III collagen (COL3A1) allele produces Ehlers-Danlos syndrome type IV in the heterozygous offspring. Am J Hum Genet. 1993;53:62–70.
Palmeri S, Mari F, Meloni I, et al. Neurological presentation of Ehlers-Danlos syndrome type IV in a family with parental mosaicism. Clin Genet. 2003;63:510–515.
Qin L, Wang J, Tian X, et al. Detection and quantification of mosaic mutations in disease genes by next-generation sequencing. J Mol Diagn. 2016;18:446–453.
Pamphlett R, Morahan JM, Yu B. Using case-parent trios to look for rare de novo genetic variants in adult-onset neurodegenerative diseases. J Neurosci Methods. 2011;197:297–301.
Hoischen A, van Bon BWM, Gilissen C, et al. De novo mutations of SETBP1 cause Schinzel-Giedion syndrome. Nat Genet. 2010;42:483–485.
Kong A, Frigge ML, Masson G, et al. Rate of de novo mutations and the importance of father’s age to disease risk. Nature. 2012;488:471–475.
Jamuar SS, Lam A-TN, Kircher M, et al. Somatic mutations in cerebral cortical malformations. N Engl J Med. 2014;371:733–743.
García-Romero MT, Parkin P, Lara-Corrales I. Mosaic neurofibromatosis type 1: a systematic review. Pediatr Dermatol. 2016;33:9–17.
Giannakudis J, Röpke A, Kujat A, et al. Parental mosaicism of JAG1 mutations in families with Alagille syndrome. Eur J Hum Genet. 2001;9:209–216.
Martínez-Quintana E, Caballero-Sánchez N, Rodríguez-González F, Garay-Sánchez P, Tugores A. Novel Marfan syndrome-associated mutation in the FBN1 gene caused by parental mosaicism and leading to abnormal limb patterning. Mol Syndromol. 2017;8:148–154.
Rohlin A, Wernersson J, Engwall Y, Wiklund L, Björk J, Nordling M. Parallel sequencing used in detection of mosaic mutations: comparison with four diagnostic DNA screening techniques. Hum Mutat. 2009;30:1012–1020.
Izawa K, Hijikata A, Tanaka N, et al. Detection of base substitution-type somatic mosaicism of the NLRP3 gene with>99.9% statistical confidence by massively parallel sequencing. DNA Res. 2012;19:143–152.
Acknowledgements
We thank all vEDS patients and their families for their kind collaboration. We acknowledge the contribution of clinicians who assessed the patients. We thank the technicians of the genetics laboratory for their contribution to the COL3A1 genetic analysis. A.L. has obtained a 3-year PhD grant from Région Ile de France. The French Medical Reference Centre for Rare Vascular Diseases (www.maladies-vasculaires-rares.fr) is supported by the French Ministry of Health. The study was supported in part by grants from Fondation pour la Recherche Médicale (FRM), the Association Française pour les Syndromes d’Ehlers Danlos (AFSED), and the Fondation GROUPAMA. This study was also supported by Assistance Publique Hôpitaux de Paris, University Paris Descartes, and INSERM. This study included vEDS patients who are now part of a national French prospective cohort (RaDiCo SEDVasc) set up by the Rare Disease Cohorts (RaDiCo) INSERM programme funded by the plan d’Investissements d’Avenir (PIA) through the Agence Nationale pour la Recherche (ANR-IO-COHO-03-01).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Disclosure
The authors declare no conflicts of interest.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Legrand, A., Devriese, M., Dupuis-Girod, S. et al. Frequency of de novo variants and parental mosaicism in vascular Ehlers–Danlos syndrome. Genet Med 21, 1568–1575 (2019). https://doi.org/10.1038/s41436-018-0356-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-018-0356-2
Keywords
This article is cited by
-
Role of Clinical Genetic Testing in the Management of Aortopathies
Current Cardiology Reports (2021)
-
The Ehlers–Danlos syndromes
Nature Reviews Disease Primers (2020)
