
Abstract
Purpose
To provide proof of concept by broadening preconception screening beyond targeted testing to inform reproductive risk in consanguineous couples.
Methods
Consanguineous couples were screened for autosomal recessive and X-linked disorders using the TruSight One panel of 4,813 genes associated with human disease.
Results
We recruited 22 couples, of whom 15 elected to have sequencing. We found four couples to be at risk of autosomal recessive disorders, including one with a child affected by Poretti–Boltshauser syndrome (a diagnosis not made prior to the study) and another previously known to carry a β-globin variant. Two couples were found to carry variants unrelated to known family history. These variants were in the genes C5orf42 (associated with Joubert syndrome and orofaciodigital syndrome) and GYS2 (associated with glycogen synthase deficiency). One known variant was not detected—a single exon deletion in FAM20C. We would not expect to identify this variant with the methodology employed. Of the four variants identified, only the β-globin variant would have been found using available commercial preconception screening panels.
Conclusion
Preconception screening of consanguineous couples for recessive and X-linked disorders using genomic sequencing is practicable, and is likely to detect many more at-risk couples than any targeted panel could achieve. A couples-based approach greatly reduces the associated analysis and counselling burden.
Similar content being viewed by others

INTRODUCTION
Genetic disease is a major cause of morbidity and mortality at all ages, but particularly in childhood. There are over 1700 known autosomal recessive (AR) disorders,1 and many more that are X-linked (XL). For most couples, the birth of an affected child is the first indication of their carrier status. Efforts to address this shortcoming by screening for carrier status in genetic disorders that are common in a given population have been strikingly successful. Examples include screening for thalassaemia in Cyprus,2 and screening for disorders with a founder effect in Ashkenazi populations.3 However, programmes of this nature are limited to targeted disorders. Even if screening is extended to tens or hundreds of conditions—as in recently available commercial panels4—only a small proportion of carrier couples are likely to be identified, because a large proportion of the overall burden of disease consists of very rare disorders.5
With the advent of increasingly inexpensive massively parallel sequencing at the level of the ‘Mendeliome’, exome and even genome sequencing could be the basis of a couple-screening programme limited only by cost-effectiveness, current knowledge and the ability to interpret results. While it would not be truly a ‘screen for everything’, it would represent a substantial advance over the status quo, in which there is usually no evidence from family history to guide targeted testing, and only general empirical population risks for a couple can be provided. The risk of having a child affected by a significant birth defect or disability is commonly quoted as 2–3% for non-consanguineous couples, or 4–6% for consanguineous couples,6 although the actual empirical risk figures vary widely between studies.7
Towards providing more comprehensive risk information for couples, we conducted a trial of preconception screening (PCS) for AR and XL disorders using the TruSight One “clinical exome” in consanguineous couples. TruSight One is a panel of 4813 genes that have been associated with human disease. Consanguineous couples were chosen as the focus for this proof-of-concept study because they are at increased risk of having children affected by AR disorders.7,8 Estimates of the burden of pathogenic recessive variants vary, but evidence from the 1000 Genomes project suggests that a typical individual carries approximately 2 such variants.9 If each person carried one variant that could be confidently assessed as pathogenic, on average one in eight couples related as first cousins would be expected to be identified as being at reproductive risk for the disorder. If each person carried 2 such variants, ~23% would be identified as being at risk for 1 or both disorders. This is calculated using the information that for a single variant there would be a 7/8 (0.875) probability that the cousin does not carry the variant. For an individual carrying 2 unlinked variants, there would thus be a 7/8 × 7/8 (i.e., 0.875 × 0.875) probability that a first cousin would carry neither variant (approximately a 77% chance). Hence, there would be a 23% chance that the cousin would carry at least one of the variants.
Although consanguineous couples are not at increased risk for XL disorders, we felt it was important to include these in the analysis because of the similar potential implications for offspring.
MATERIALS AND METHODS
This study was approved by the South Eastern Sydney Local Health District Human Research Ethics Committee (reference number 14/026)
Recruitment
Subjects were recruited via participating clinical genetics units. All but one couple were from Liverpool Hospital, which provides services to an ethnically diverse population group in south-western Sydney, many of whom are Arabic speaking. The remaining couple were recruited from the Royal Hospital for Women in eastern Sydney. Invitations were sent to couples who previously consulted a clinical geneticist or genetic counsellor for any indication, and who were thought likely to be eligible to participate in the study. Couples were eligible if they were related at least as second cousins—equivalent to a coefficient of inbreeding (F) in their progeny of 0.0156—and were planning further children at the time of initial contact. A current ongoing pregnancy was an exclusion criterion. Health interpreters were used as required. In addition, study documentation was translated into Arabic.
Consent for testing
Consent was obtained by one of two investigators (E.P.K. or K.B.-S.)—a clinical geneticist and senior genetic counsellor, respectively. The consent process included discussion of the possibility of incidental findings, as well as an explicit statement of the small risk that a variant might be incorrectly classified as pathogenic. If this were to occur, couples might be wrongly identified as being carriers. There was also discussion of the limitations of the test, and the fact that there would remain a residual risk after screening that could not be quantified. As part of a concurrent qualitative study, interviews with the couples were recorded, exploring the understanding of and attitudes towards issues related to consanguinity before obtaining consent for testing. All couples enrolled in the study gave consent for return of the results of PCS.
Genomic testing and analysis
Genomic DNA was extracted from samples of each partner’s peripheral blood. The TruSight One panel was run on a NextSeq 500 instrument (Illumina) at the Ramaciotti Centre, University of New South Wales. Alignment and variant calling were performed in BaseSpace using the Burrows–Wheeler Aligner and Genome Analysis Toolkit (Burrows–Wheeler Aligner enrichment application). Variant-effect prediction and initial filtering were performed using the Seave genomic analysis platform developed at the Garvan Institute with the GEMINI (Genome MINIng) version 0.19.1 programme as the underlying platform.10
Variants were filtered according to the following criteria. For XL genes, only heterozygous variants present in the female partner were considered. For genes associated with AR disorders, variants were considered only if one of the following applied: (1) both partners were heterozygous for the same variant; or (2) each partner was heterozygous for a different variant in the same gene, with both variants meeting the other filtering criteria. Variants with minor allele frequency in the Genome Aggregation Database or 1000 Genomes of >0.01 were excluded, as were variants present in 3 or more couples from the cohort. Variants in genes without a known human Mendelian disease association were excluded. The remaining variants were hand curated. For loss-of-function variants, consideration was given to the strength of evidence for an association between the gene and human disease, whether loss-of-function variants were a known mechanism of disease and whether all or only some known splice variants of the gene would be affected by the variant.
The analysis was performed blinded to previously collected clinical information about the family. Once the analysis had been completed and a decision had been made regarding which variants could be returned to families, the information was unblinded. Variants that had not been independently identified in the family were confirmed using Sanger sequencing.
RESULTS
Recruitment
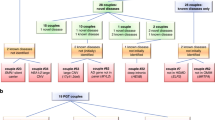
We invited couples to explore their interest in obtaining risk information for having an affected child. We received responses from 22 couples, 21 of whom consented to participate in the concurrent qualitative study. After discussion of the nature of the study, 15 couples consented to genomic testing.
One couple were related as second cousins (F = 0.0156). The remainder were related as first cousins (F = 0.0625). Mean ages were 27 years (range 20–39 years) for women and 34 years (range 25–46 years) for men. The couples had had a median of two previous children (three couples had no children; no family had more than three). Seven couples already had a child with a significant health problem with no known cause (and that could have been due to a recessive disorder). Three couples had a family history of such a disorder. The remainder had neither affected children nor a relevant family history. At the time of recruitment, two of the couples were known to be carriers of AR disorders: β-thalassaemia and Raine syndrome, respectively.
Molecular genetic findings
After filtering, a median of 7 variants (range 1–14) remained for further assessment. Of these, three couples had a single loss-of-function variant (present in both partners), with the remainder being missense variants. No carriers of XL disorders were identified, and no couples were found to be heterozygous for different pathogenic variants in the same gene. In four couples, both partners were found to carry the same pathogenic variant, and were thus identified as being at risk of having a child affected by an AR disorder (Table 1).
DISCUSSION
The disorders for which the couples were found to be at risk are illustrative of the variety of AR conditions encountered, and the limited value of panels restricted to relatively common conditions.
Variants identified
C5orf42
Variants in C5orf42 have been reported in association with Joubert syndrome11 and orofaciodigital syndrome,12—AR neurodevelopmental syndromes with overlapping features. Loss of function is an established mutational mechanism.11,12,13 Although the variant identified, NM_023073.3(C5orf42):c.3792del (p.Val1265Phefs*6) has not been reported previously, it is predicted to result in a frameshift in exon 21 of 53, with the introduction of a premature stop codon after 6 amino acids. The resulting transcript is thus likely to be targeted for nonsense-mediated decay. More distally located nonsense and frameshift variants have been reported in affected individuals.11 There are no known transcripts that do not include exon 21.
GYS2
The GYS2 gene encodes glycogen synthase—an essential enzyme for glycogen synthesis. Glycogen synthase deficiency is characterized by fasting intolerance with morning hypoglycaemia14 and, if untreated, there can be long-term neurological sequelae.15 Diagnosis can be difficult, but once diagnosed, treatment is relatively simple. The variant NM_021957.3(GYS2):c.736 C > T (p.Arg246Ter) creates a premature stop codon in exon 5 of 16, with the resulting transcript likely to be targeted for nonsense-mediated decay. The variant has been reported previously in the homozygous state in an affected individual15 (OMIM reference 1385710001; ClinVar RCV000017427.28).
HBB
Variants in HBB result in a range of haemoglobinopathies, including β-thalassaemia and sickle cell anaemia. NM_000518.4(HBB):c.92 G > A (p.Arg31Lys) is a previously reported variant16 and had already been identified in the couple as the result of routine screening.
LAMA1
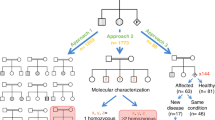
Variants in LAMA1 have been associated with Poretti–Boltzhauser syndrome, which is characterized by variable intellectual disability, cerebellar abnormalities and retinal dystrophy.17,18 Variants reported to date have been predicted to result in loss of function. Couple C002 have a son who has intellectual disability, cerebellar hypoplasia and a retinal disorder, which had been described as ocular albinism. Shortly after the couple were enrolled in this study, a clinical geneticist ordered clinical exome sequencing on the affected child, which revealed homozygosity for the NM_005559.3(LAMA1):c.3597del (p.Asp1200Thrfs*38) variant. On review with this information, a clinical diagnosis of Poretti–Boltzhauser syndrome was confirmed. This couple had previously had a pregnancy affected by Raine syndrome, making them the only couple in the cohort known to be at risk for two separate conditions. This was also the only instance in which carrier status was identified that proved relevant to a hitherto undiagnosed condition in the family.
The current guidelines from professional groups, such as the American College of Medical Genetics and Genomics19 and the European Society of Human Genetics20 recommend caution in the implementation of expanded PCS. In particular, they advocate careful selection of disorders for inclusion in panels. Arguments advanced for such a measured approach are intended to consider and address the detection of couples who are carriers of relatively mild disorders, the possibility of incidental findings, possible negative psychological impacts on the large number of individuals who are found to be carriers, and the difficulties of variant analysis and classification. These are real issues that require consideration. However, in our view, the potential disadvantages are outweighed by the significant shortcomings of targeted panels; in particular, the large number of disorders they fail to detect, and thus the large proportion of at-risk couples they fail to assist.
A recent Committee Opinion from the American College of Obstetricians and Gynecologists21 is supportive of expanded carrier screening, but stipulates a carrier frequency of at least 1 in 100. However, carrier frequencies are seldom available for rare disorders, which often are restricted to particular families and sub-communities,8,22 and the recommended threshold seems arbitrary and hard to justify. If it is possible to screen for a disorder with a carrier frequency of 1 in 110, or even 1 in 1000, why not do so? The concerns regarding the identification of mild disorders or incidental findings can readily be addressed at the filtering step, by bioinformatically excluding a list of genes for which these issues are a consideration.
One potential technical obstacle to screening for very large numbers of conditions is the requirement to assess large numbers of variants for pathogenicity. However, as we demonstrate here, the combined analysis of the data for both partners dramatically reduces the number of variants that need to be assessed. For first-cousin couples, nearly seven out of eight autosomal variants can be filtered out without further analysis; only the one out of eight variants that are identical by descent and the small number that are shared by chance require detailed review. If the same approach was applied to unrelated couples, it is likely that only a small number of variants would require assessment. None of the 15 couples in this study had any shared variants that passed filtering and were not identical by descent; extrapolating from this sample, in non-consanguineous couples, the workload required for variant analysis would be minimal.
The approach also means that carrier individuals who are deemed not to be at risk of having affected offspring within the limitations of the couple testing will not be identified. This reduces the counselling requirement in follow-up, removes considerations of cascade testing (which would be unreasonably burdensome for a test that would be likely to identify almost every tested individual as being a carrier) and allows the application of resources to those who are most at need (i.e., couples at risk of having children affected by AR and XL disorders). There are potential disadvantages to the couples-based approach. One is the loss of the opportunity to conduct cascade screening for variants not analysed and reported. While this may not be desirable for very rare conditions, it could be argued that it is problematic for more common conditions, such as cystic fibrosis. Wide access to couples-based screening would obviate this concern. Another issue is the inevitability that some couples will separate and subsequently find new partners. For this reason, education regarding the applicability of the approach only to a specific couple, with the need for additional testing and analysis where there is a new partner, will need to form an important part of pre- and post-test counselling.
Based on earlier studies,9 which suggest a detectable burden of recessive variants of around one to two per individual, we predicted that we would identify at least one in eight couples as being at risk. In fact, 4 of 15 couples were found to carry a variant that placed them at risk of having a child affected by an AR disorder. Of these four variants, only one (the HBB variant) could have been expected to be identified by one of the panels currently available commercially. However, it would also be expected to be detected by routine screening based on ethnicity, as was the case.
Two of the couples (close to the predicted one in eight) had variants identified that were unrelated to family history. One of these, the GYS2 variant, demonstrates a less-emphasized potential benefit of expanded PCS. Glycogen synthase deficiency is a disorder that can have severe consequences if undiagnosed, including hypoglycaemic seizures, developmental delay and failure to thrive. The diagnosis is often difficult to make, but treatment is relatively straightforward. If couples perceived that the disorder was too mild for them to seek PGD or prenatal diagnosis, the information would still be valuable because of the likely substantial benefits for an affected child from early diagnosis.
Limitations and challenges
The approach described here has limitations. Utilizing exome or, even better, genome sequencing might have identified additional at-risk couples compared with the TruSight One ‘clinical exome’ we employed. For example, we failed to detect the known variant in FAM20C in one couple, who previously had a pregnancy affected by Raine syndrome. The FAM20C variant in question was a single exon deletion that was not detectable using the methods we employed. genome sequencing in particular would have been more likely to detect this variant (and potentially other pathogenic copy-number variants).
Recruitment via a clinical genetics service has the potential to introduce bias, due to an over-representation of couples with children affected by genetic conditions compared with a population-based sample. In practice, only the LAMA1 variant was relevant to the original reason the couples were referred to the genetics service. Thus, our results are likely to be relevant to population-based screening of consanguineous couples.
A priority of our study was minimizing the risk of wrongly advising a couple that they were at risk, which required a very conservative approach to variant curation. In this context, the evaluation of missense variants is challenging, whereas it is more straightforward to assess loss-of-function variants, even though considerable caution is still required. We anticipate that missense variants could only be returned to patients where there is strong documented evidence for pathogenicity. For loss-of-function variants, attention must be paid to the position of the variant within the gene, including the possibilities that: (1) a variant at the 3′ end of a gene may escape nonsense-mediated decay; and (2) alternate splicing might result in the production of a functional protein, from isoforms that do not include the region affected by the variant.
Clinical utility
The couples who consented to testing were seeking increased efficacy in their reproductive risk information for future pregnancies. The importance of the clinical utility of this testing for all the consanguineous couples interviewed is considered in a separate paper (manuscript submitted) describing the qualitative component of the study.
Conclusion
Despite the identified limitations of the technology used, the present study confirms that there is a large potential benefit to this approach to PCS, and we argue that these benefits substantially outweigh the potential problems: the concept has been proven and it has perceived clinical utility. We propose that an optimal future screening programme would consist of couples-based genome sequencing, with copy-number variation analysis included, as well as fragile X and spinal muscular atrophy testing (unless genome sequencing can be validated to remove the need for a separate test for spinal muscular atrophy and/or fragile X).
It is time to embrace the potential of the technology that is already in our hands, and move beyond the panel.
REFERENCES
Sulem P, Helgason H, Oddson A, et al. Identification of a large set of rare complete human knockouts. Nat Genet 2015;47:448–452.
Angastiniotis M, Kyriakidou S, Hadjiminas M. How thalassaemia was controlled in Cyprus. World Health Forum 1986;7:291–297.
Bach G, Tomczak J, Risch N, et al. Tay–Sachs screening in the Jewish Ashkenazi population: DNA testing is the preferred procedure. Am J Med Genet 2001;99:70–75.
Xue Y, Ankala A, Wilcox W, et al. Solving the molecular diagnostic testing conundrum for Mendelian disorders in the era of next-generation sequencing: single-gene, gene panel, or exome/genome sequencing. Genet Med 2014;17:444–451.
Lacaze P, Millis N, Fookes M, et al. Rare disease registries: a call to action. Int Med J 2017;47:1075–1079.
Hamamy H, Antonarakis SE, Cavalli-Sforza LL, et al. Consanguineous marriages, pearls and perils: Geneva International Consanguinity Workshop Report. Genet Med 2011;13:841–847.
Bittles AH. Consanguinity and Morbidity in Early Life. Consanguinity in Context. Cambridge: Cambridge University Press; 2012. 136–161.
Bittles A. Consanguinity, genetic drift, and genetic diseases in populations with reduced numbers of founders. In: Speicher M, Motulsky A, Antonarakis S, editors. Vogel and Motulsky’s Human Genetics. Berlin: Springer; 2010. 507–528.
Xue Y, Chen Y, Ayub O, et al. Deleterious- and disease-allele prevalence in health individuals: insights from current predictions, mutation databases, and population-scale resequencing. Am J Hum Genet 2012;91:1022–1032.
Paila U, Chapman BA, Kirchner R, et al. GEMINI: integrative exploration of genetic variation and genome annotations. PLoS Comput Biol 2013;9:e1003153.
Srour M, Schwartzentruber J, Hamdan FF, et al. Mutations in C5ORF42 cause Joubert Syndrome in the French Canadian population. Am J Hum Genet 2012;90:693–700.
Lopez E, Thauvin-Robinet C, Reversade B, et al. C5orf42 is the major gene responsible for OFD syndrome type VI. Hum Genet 2014;133:367–377.
Wentzensen I, Johnston J, Keppler-Noreuil K, et al. Exome sequencing identifies novel mutations in c5orf42 in patients with Joubert syndrome with oral–facial–digital anomalies. Hum Genome Var 2015;2:1–3.
Weinstein DA, Correia CE, Saunders AC, et al. Hepatic glycogen synthase deficiency: an infrequently recognized cause of ketotic hypoglycaemia. Mol Genet Metab 2006;87:284–288.
Orho M, Bosshard NU, Buist NR, et al. Mutations in the liver glycogen synthase gene in children with hypoglycaemia due to glycogen storage disease type 0. J Clin Invest 1998;102:507–515.
Al-Gazali L, Ali BR Mutations of a country: a mutation review of single gene disorders in the United Arab Emirates (UAE). Hum Mutat 2010;31:505–520.
Poretti A, Hausler M, von Moers A, et al. Ataxia, intellectual disability, and ocular apraxia with cerebellar cysts: a new disease? Cerebellum 2014;13:79–88.
Aldinger KA, Mosca SJ, Tetreault M, et al. Mutations in LAMA1 cause cerebellar dysplasia and cysts with and without retinal dystrophy. Am J Hum Genet 2014;95:227–234.
Grody WW, Thompson BH, Gregg AR, et al. ACMG position statement on prenatal/preconception expanded carrier screening. Genet Med 2013;15:482–483.
Henneman L, Borry P, Chokoshvili D, et al. Responsible implantation of expanded carrier screening. Eur J Hum Genet 2016;24:e1–e12.
American College of Obstetricians and Gynecologists Carrier screening in the age of genomic medicine. Committee Opinion No. 690. Obstet Gynecol 2017;129:e35–e40.
Corry PC Consanguinity and the prevalence patterns of inherited disease in the UK Pakistani community. Hum Hered 2014;77:207–216.
ACKNOWLEDGEMENTS
This study was funded by a grant from the Apex Foundation for Research into Intellectual Disability. We acknowledge T. Le of NSW Health Pathology for completing the confirmatory Sanger sequencing of the detected pathogenic variants.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
DISCLOSURE
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Kirk, E.P., Barlow-Stewart, K., Selvanathan, A. et al. Beyond the panel: preconception screening in consanguineous couples using the TruSight One “clinical exome”. Genet Med 21, 608–612 (2019). https://doi.org/10.1038/s41436-018-0082-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-018-0082-9
Keywords
This article is cited by
-
Challenges for the implementation of next generation sequencing-based expanded carrier screening: Lessons learned from the ciliopathies
European Journal of Human Genetics (2023)
-
Diagnostic yield of clinical exome sequencing as a first-tier genetic test for the diagnosis of genetic disorders in pediatric patients: results from a referral center study
Human Genetics (2022)
-
Carrier detection probabilities for autosomal recessive variants in unrelated and consanguineous couples — an evaluation of the 86 genes of the ACMG ‘Tier 3’ panel
Journal of Community Genetics (2022)
-
Parental exome analysis identifies shared carrier status for a second recessive disorder in couples with an affected child
European Journal of Human Genetics (2021)
-
Challenges and Controversies in the Genetic Diagnosis of Hereditary Spastic Paraplegia
Current Neurology and Neuroscience Reports (2021)
