
Abstract
Purpose
The reclassification of genetic variants poses a significant challenge for laboratories and clinicians. Variant review has resulted in the reclassification of variants of unknown significance as well as the reclassification of previously established pathogenic and likely pathogenic variants. These reclassifications have the potential to alter the clinical management of patients with hereditary cancer syndromes.
Methods
Results were reviewed for 1694 patients seen for hereditary cancer evaluation between August 2012 and May 2017 to determine the frequency and types of variant reclassification. Patients with reclassifications with high potential for impact were monitored for alterations in organ surveillance, prophylactic surgery, and cascade testing.
Results
One hundred forty-two variants were reclassified representing 124/1694 (7.3%) patients; 11.3% of reclassifications (16/142) had a high potential for clinical impact with 94% (15/16) altering clinical management of patients with 56% (9/16) changing multiple areas of management.
Conclusion
While reclassifications are rare, the impact on clinical management is profound. In many cases, patients with downgraded pathogenic/likely pathogenic variants had years of unnecessary organ surveillance and underwent unneeded surgical intervention. In addition, cascade testing misidentified those at risk for developing cancers, thereby altering the management across generations. The frequency and types of alterations to clinical management highlight the need for timely variant reclassification.
Similar content being viewed by others


Introduction
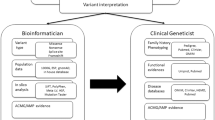
The integration of next-generation sequencing (NGS) into clinical medicine has revolutionized genetic testing in the diagnosis, prognosis, and clinical management for a range of inherited disorders, including hereditary cancer syndromes.1,2,3,4,5,6 Testing expansion has yielded increases in diagnostic yield1,7,8,9 but also a substantial increase in reported variants of unknown significance (VUS).6,10 These variants are not clinically diagnostic of a cancer syndrome, but can still present significant challenges in the counseling of patients and their families regarding appropriate management for cancer risks.
Variant interpretation guidelines issued by the American College of Medical Genetics and Genomics and the Association for Molecular Pathology recommend that testing laboratories have policies regarding the re-review of genetic data.11 These guidelines do not explicitly define the mechanism for re-review of variant data. Commercial laboratories report that this is typically satisfied through systematic review of VUS or more commonly reinterpretation of variant classifications upon provider request. In either case, these recommendations suggest that the testing laboratory has a responsibility to report new findings to the provider, allowing them to take appropriate and timely clinical action.12
Recent reports have shown that reclassifications of hereditary cancer variants often result in changing a VUS to benign or likely benign; however, a small percentage of variant reclassifications have been shown to result in the new diagnosis or a diagnostic revision of a hereditary cancer.13 However, to date, no studies have examined the direct clinical impact of these variant reclassifications on affected patients. In this study we examine the impact of variant reclassification on clinical management of patients at risk for a hereditary cancer syndrome.
Materials and methods
Genetic testing lab reports were reviewed from 1694 patients tested at the Vanderbilt Hereditary Cancer Clinic at the Vanderbilt-Ingram Cancer Center between August 2012 and May 2017. Germline testing was submitted to one of four commercial laboratories for the clinical diagnosis of a suspected hereditary cancer syndrome. The genes tested were dependent on the patient’s personal and family history of cancer and current testing guidelines.
Amended reports with variant reclassifications were provided by the original testing laboratory. For the purpose of this study, no variants were considered reclassified unless the testing laboratory issued an amended clinical report. Therefore, variants tested by multiple laboratories but not reclassified by the original testing laboratory may not have been included in this study. While reclassifications resulting from familial studies were included other provider-requested reclassifications were excluded from this study.
To determine the clinical impact of variant reclassification, the medical records at time of initial classification and upon reclassification were reviewed. Changes in recommendation in three areas were noted: (1) frequency or type of organ-specific surveillance, (2) cascade testing for family members, and (3) prophylactic surgical interventions. A reclassification with an alteration in at least one of these areas was considered as having a clinical impact in patient management.
Results were tracked using a 21 Code of Federal Regulations (CFR) Part 11, Federal Information Security Management Act (FISMA), and Health Insurance Portability and Accountability Act of 1996 (HIPAA)-compliant Research Electronic Data Capture (RedCap) database.14 This study was approved by the Vanderbilt Institutional Review Board (141620).
Results
At least one VUS was identified in 25.4% (431/1694) of patients, with 7.0% (119/1694) of patients having more than one VUS reported. Deleterious (pathogenic/likely pathogenic) variants were reported in 20.0% (338/1694) of patients, with < 1% (14/1694) of patients having more than one deleterious variant reported. 2.5% (42/1694) of patients had both a deleterious variant and at least one VUS reported. No reportable variants (pathogenic, likely pathogenic, or VUS) were identified in 59.1% (1002/1694) of patients (Table 1).
Of all clinical reports, 7.3% (124/1694) were amended with an upgraded or downgraded interpretation. Surprisingly, 3.7% (13/351) of pathogenic or likely pathogenic variants were reclassified. Reclassification reports were most commonly issued for VUS with 21.7% (129/592) of reported VUS undergoing reclassification (Table 1). The mean time to a reissued clinical report was 519.2 days (13–1799 days). At the completion of the study 78% (463/592) of VUS had not been reclassified with an elapsed mean time of 778.5 days from report.
Of 142 variants, 16 (11.3%), representing 16 patients from 13 families, resulted in the highest potential to alter clinical management. The reissued reports included downgrading a pathogenic or likely pathogenic variant to a VUS in 6 patients (Table 2), and upgrading VUS to pathogenic or likely pathogenic in 10 patients (Table 3). Of all reclassifications, 7.1% resulted in changes in confidence (i.e., likely pathogenic to pathogenic) and 81.6% (116/142 variants) resulted in a VUS being downgraded to benign or likely benign.
Changes in management were noted for 95% (15/16) of these high-risk patients with 56% (9/16) resulting in alterations to multiple areas of management (Table 2, Table 3). The most common change in clinical management was altered recommendation for cascade testing for family members occurring in 75% (12/16) of the patients. Sixty-nine percent (11/16) of patients had changes in recommendations for frequency or types of organ surveillance warranted. Thirty-one percent (5/16) of patients had altered recommendations for a combination of cascade screening and organ surveillance. Twenty-five percent (4/16) of patients had changes in recommendation for surgical interventions. While alterations in all three categories occurred in 19% (3/16) patients.
The six patients with deleterious variants downgraded to VUS included five different variants in genes BRCA1, BRCA2, TP53, and CHEK2 (Table 2) and led to alterations in clinical management. One patient was lost to follow-up due to death prior to reclassification.
The proband for family 1 was a 53-year-old Caucasian female with a personal history of breast cancer at 53 years of age and a family history remarkable for breast, colon, sarcoma, and melanoma in at least three generations. Testing identified a pathogenic TP53 variant (p.Met237Val) associated with the diagnosis of Li–Fraumeni syndrome. Recommendations for management were made according to National Comprehensive Cancer Network (NCCN) guidelines, and provider recommendations, including surgical recommendation to avoid radiation therapy due to increased risk for radiation-induced second primary tumors, organ surveillance through yearly whole-body magnetic resonance image (MRI) scans, and cascade testing for all family members regardless of age. The patient opted to have bilateral mastectomies and prophylactic bilateral salpingo-oophorectomy (BSO) based on surgical provider recommendations, initiated additional organ surveillance, and recommended to family members they be tested to determine their own risk. Note that the BSO recommendations are outside of NCCN guidelines. A reclassification report was issued downgrading a pathogenic variant to a VUS 943 days after original report. Upon reclassification, clinical management was altered to stop yearly whole-body MRI. Prophylactic surgery had been performed. Family members who tested negative for this variant were re-evaluated and recommendations reverted to screening appropriate for an individual with a family history of breast cancer.
The proband from family 3 was an 86-year-old Caucasian female with breast cancer diagnosed at 46 years of age and a family history concerning for a hereditary breast and ovarian cancer syndrome (HBOC). A pathogenic BRCA2 varaint (p.Cys3233Trpfs*15) was initially identified and HBOC was confirmed. The proband elected not to have risk-reducing surgery due to advanced age and residual lifetime risk. According to NCCN guidelines, alterations to breast surveillance were recommended and cascade testing offered. Cascade testing identified one daughter positive and three additional children as negative for the BRCA2 variant. A daughter who tested negative had a personal history of ovarian cancer and elected to perform a risk-reducing bilateral mastectomy. This variant was initially classified as pathogenic as it results in a frame-shift mutation, and while at the 3’ end of the gene, the notation of other pathogenic variants (p.Tyr3308*) supported this initial classification.15 Subsequently, this variant was seen in trans with other known pathogenic BRCA2 variants in patients who did not display features of Fanconi anemia and this variant did not segregate with breast and ovarian cancer in families.16 However, some contribution of this variant on disease cannot be entirely excluded.17 This variant is considered a “special interpretation variant” and reported as either likely benign or as a VUS. Upon reclassification, the clinical management reverted back to screening based on family history. While this did not impact the clinical care of the proband, it had a significant impact on her daughters. Recommendations for risk-reducing bilateral mastectomy would have been altered. Family members who initially did not carry the BRCA2 variant now require additional screening and clinical follow-up based on family history.
The proband of family 4 was a 70-year-old Caucasian female with multiple synchronous primaries of the thyroid, head, and neck diagnosed at the age of 66, a history of greater than 10 adenomatous colon polyps over her lifetime, and a family history of breast and colon cancer. A CHEK2 pathogenic variant was initially identified and the patient was counseled for an increased risk for breast, ovarian, and possibly colon cancer. While the clinical guidelines for CHEK2 mutations are currently under consideration, recommendations were made for altered organ surveillance and cascade testing for family members. Upon re-review of pertinent literature, the laboratory downgraded this variant to a VUS. The functional data used as evidence to classify this as a pathogenic variant was deemed insufficient.18 The physiological relevance of this CHEK2 variantn is currently unknown. Clinical recommendations for this patient were altered, including reducing organ surveillance based on family history of cancer, and altered recommendation to perform cascade screening on only affected members of the family, to provide further clarification on variant segregation.
The 10 patients with variants upgraded to likely pathogenic or pathogenic included eight different variants in HNF1A, MSH6, BRCA1, SDHD, and PMS2 (Table 3) and led to alterations in clinical management. The consequence of delayed diagnosis for these patients was assessed through chart review. None of the described patients reported any new cancers between initial classification and reclassification. However, some patients may have had medical care outside of the medical center limiting further clinical review.
Discussion
This study highlights that variant reclassification can lead to altered clinical recommendations for patients and their families. We found that 12% of reclassifications resulted in a change in major reporting category with a potential to impact patient management. Notably, our study supports a recent publication that found a similar proportion of variant reclassifications.13
While the majority of these variants led to clinical recommendation fitting a diagnosis of a hereditary cancer syndrome, an unexpected number of patients were issued revisions on their diagnosis of hereditary cancer. For patients with a new diagnosis of a hereditary cancer, management recommendation for enhanced organ surveillance, prophylactic surgery, and cascade testing have the potential to reduce risk or more readily identify and treat new cancers. To our knowledge none of the patients described herein reported new cancers during the reclassification period. Additionally, unexpected clinical implications resulted from the revised diagnosis of a hereditary cancer in some patients. Such reclassifications resulted in a reduction of organ surveillance, reducing the impact of invasive procedures on the patient and potentially reducing the burden of clinical management team who perform these procedures. Surgical recommendations are often considered at time of diagnosis, and therefore at reclassification prophylactic surgery may have been already performed.
Unexpectedly, these reclassified variants had additional impacts on the healthcare needs of the patient’s family. When a pathogenic variant is downgraded, its role in cancer predisposition is no longer considered definitive; therefore family member variant status is no longer relevant in determining cancer risk. This may reduce or increase an individual’s relative risk for developing cancer. For example, a daughter in family 3 tested ”negative” for the initial BRCA2 variant, so her risk for developing breast or ovarian cancer would have been that of the general population. However, reclassification to a VUS alters risk estimates to reflect her familial risk for developing certain cancers. In other words, her initial negative report is now a VUS, and therefore no longer relevant to estimating her cancer risks. The converse is true of the daughter who tested positive for the variant. Her relative risk for the indicated cancers is reduced and should now be based on family history.
The clinical impact of the length of time to reclassification is difficult to determine. It is suspected that lengthy waits for reclassifications may result in inappropriate care for the small percentage of patients who have a hereditary cancer syndrome. Furthermore, as time goes by, patients and their families can be lost to follow-up due to relocation or death. This places a substantial burden on the clinical team to track down these patients to properly counsel them regarding their updated risk. The necessity for laboratories to report downgraded VUS to clinicians and clinicians to patients is an important topic of current debate. However, while the clinical impacts of these reclassifications are negligible, as shown here, the social and psychological impacts should not be overlooked and need further investigation.
Given the numbers of VUS reported, the burden of periodic reclassification on clinical laboratories is considerable and potentially unsustainable. To mitigate this burden, providers should submit de-identified variants to an online database that tracks and updates classifications. If a variant is flagged as reclassified, the provider should request a review and an amended report should be issued quickly if new evidence warrants and the provider can reasonably recontact the patient.
Lastly, this study highlights challenges faced by laboratories with evolving guidelines in variant interpretation. First, it was noted that many laboratories revamped internal variant interpretation algorithms upon publication of the 2015 guidelines resulting in reclassification based on altered significance to available evidence. Second and most commonly, variants were reclassified due to new proprietary evidence not available to all testing laboratories. The proprietary nature of internal variant databases makes evaluation of data by outside parties impossible. This data should be incorporated into publicly available databases, such as ClinGen, to allow for data aggregation and quicker resolution of rare variants. Ideally, the data should include pathogenic and benign variants. Third, amended reports should contain updated references including both new and reinterpreted clinical or functional studies supporting reclassification. While the guidelines incorporate functional data, the quality and clinical conclusions of these studies must be carefully interpreted by a genetics professional, especially in the absence of additional clinical data. To date, computational tools fall far short, requiring careful manual review and re-review of all published reports. Fourth, reports were reissued due to classification contradictions between somatic and germline variants. The level of evidence for classifying variants as disease causing in tumors differs from what is recommended for pathogenic germline classification.19 This problem is particularly profound in evaluating TP53 variants as many occur in somatic hotspots within the DNA binding domain and have been observed across a variety of tumor types (COSMIC).20 These variants are much more rare in germline tissues; therefore, the data to support a pathogenic classification is not otherwise available. Finally, the existence of “special interpretation variants,” those having strong evidence to suggest pathogenicity but additional contradictory data, further complicates clinical evaluation. While this class of variants is rare,17 the potential for clinical impact warrants additional considerations. Such cases highlight the need for continued evaluation of genetic data regardless of initial classification. Therefore, the variant interpretation must be viewed in light of the clinical evaluation of the patient and family when determining management recommendations.
Over the course of this study reclassification was found to be uncommon, but as demonstrated, could have a substantial clinical impact on a case-by-case basis. The discovery of rare variants will undoubtedly continue to rise and the protracted length of time for reclassification suggests that laboratories need more comprehensive policies on re-review of data regardless of provider request. While this study highlights the clinical impact of reclassification, the social and psychological impacts on patients need to be considered in policies regarding reissue of clinical reports.
References
Ricker C, Culver JO, Lowstuter K et al. Increased yield of actionable mutations using multi-gene panels to assess hereditary cancer susceptibility in an ethnically diverse clinical cohort. Cancer Genet. 2016;209:130–7.
Hermel DJ, McKinnon WC, Wood ME et al. Multi-gene panel testing for hereditary cancer susceptibility in a rural familial cancer program. Fam Cancer. 2017;16:159–66.
Mauer CB, Pirzadeh-Miller SM, Robinson LD et al. The integration of next-generation sequencing panels in the clinical cancer genetics practice: an institutional experience. Genet Med. 2014;16:407–12.
Yorczyk A, Robinson LS, Ross TS. Use of panel tests in place of single gene tests in the cancer genetics clinic. Clin Genet. 2015;88:278–82.
Nagy R, Sweet K, Eng C. Highly penetrant hereditary cancer syndromes. Oncogene. 2004;23:6445–70.
LaDuca H, Stuenkel AJ, Dolinsky JS et al. Utilization of multigene panels in hereditary cancer predisposition testing: analysis of more than 2,000 patients. Genet Med. 2014;16:830–7.
Cragun D, Radford C, Dolinsky JS et al. Panel-based testing for inherited colorectal cancer: a descriptive study of clinical testing performed by a US laboratory. Clin Genet. 2014;86:510–20.
Tung N, Battelli C, Allen B et al. Frequency of mutations in individuals with breast cancer referred for BRCA1 and BRCA2 testing using next-generation sequencing with a 25-gene panel. Cancer. 2015;121:25–33.
Gallego CJ, Shirts BH, Bennette CS et al. Next-generation sequencing panels for the diagnosis of colorectal cancer and polyposis syndromes: a cost-effectiveness analysis. J Clin Oncol. 2015;33:2084–91.
Pugh TJ, Kelly MA, Gowrisankar S et al. The landscape of genetic variation in dilated cardiomyopathy as surveyed by clinical DNA sequencing. Genet Med. 2014;16:601–8.
Richards S, Aziz N, Bale S et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24.
Hirschhorn K, Fleisher LD, Godmilow L et al. Duty to re-contact. Genet Med. 1999;1:171–2.
Macklin S, Durand N, Atwal P et al. Observed frequency and challenges of variant reclassification in a hereditary cancer clinic. Genet Med. 2017;20:346–50.
Harris PA, Taylor R, Thielke R et al. Research electronic data capture (REDCap)--a metadata-driven methodology and workflow process for providing translational research informatics support. J Biomed Inform. 2009;42:377–81.
Biswas K, Das R, Eggington JM et al. Functional evaluation of BRCA2 variants mapping to the PALB2-binding and C-terminal DNA-binding domains using a mouse ES cell-based assay. Hum Mol Genet. 2012;21:3993–4006.
Howlett NG, Taniguchi T, Olson S et al. Biallelic inactivation of BRCA2 in Fanconi anemia. Science. 2002;297:606–9.
Rosenthal ET, Bowles KR, Pruss D et al. Exceptions to the rule: case studies in the prediction of pathogenicity for genetic variants in hereditary cancer genes. Clin Genet. 2015;88:533–41.
Bell DW, Kim SH, Godwin AK et al. Genetic and functional analysis of CHEK2 (CHK2) variants in multiethnic cohorts. Int J Cancer. 2007;121:2661–7.
Li MM, Datto M, Duncavage EJ et al. Standards and guidelines for the interpretation and reporting of sequence variants in cancer: a joint consensus recommendation of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists. J Mol Diagn. 2017;19:4–23.
Weitzel JN, Chao EC, Nehoray B, et al. Somatic TP53 variants frequently confound germ-line testing results. Genet Med. 2017; doi:10.1038/gim.2017.196 PMID 29189820.
Acknowledgements
We thank the genetic counselors, advanced practice nurses, and the patients and their families who were seen at the Vanderbilt Hereditary Cancer Clinic at Vanderbilt-Ingram Cancer Center for participating in this study. G.L.W. is supported through the Vanderbilt-Ingram Cancer Center (VICC).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
DISCLOSURE
The authors declare no conflicts of interest.
Rights and permissions
About this article

Cite this article
Turner, S.A., Rao, S.K., Morgan, R.H. et al. The impact of variant classification on the clinical management of hereditary cancer syndromes. Genet Med 21, 426–430 (2019). https://doi.org/10.1038/s41436-018-0063-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-018-0063-z
Keywords
This article is cited by
-
Landscape of germline pathogenic variants in patients with dual primary breast and lung cancer
Human Genomics (2023)
-
Exploring the impact of the reclassification of a hereditary cancer syndrome gene variant: emerging themes from a qualitative study
Journal of Community Genetics (2023)
-
BRCA1 Norway: comparison of classification for BRCA1 germline variants detected in families with suspected hereditary breast and ovarian cancer between different laboratories
Familial Cancer (2022)
-
An ethical analysis of divergent clinical approaches to the application of genetic testing for autism and schizophrenia
Human Genetics (2022)
-
Endophenotype effect sizes support variant pathogenicity in monogenic disease susceptibility genes
Nature Communications (2022)
