
Abstract
Purpose
The Veterans Health Administration (VHA) Clinical Pharmacogenetics Subcommittee is charged with making recommendations about whether specific pharmacogenetic tests should be used in healthcare at VHA facilities. We describe a process to inform VHA pharmacogenetic testing policy.
Methods
After developing consensus definitions of clinical validity and utility, the Subcommittee identified salient drug–gene pairs with potential clinical application in VHA. Members met monthly to discuss each drug–gene pair, the evidence of clinical utility for the associated pharmacogenetic test, and any VHA-specific testing considerations. The Subcommittee classified each test as strongly recommended, recommended, or not routinely recommended before drug initiation.
Results
Of 30 drug–gene pair tests reviewed, the Subcommittee classified 4 (13%) as strongly recommended, including HLA-B*15:02 for carbamazepine-associated Stevens–Johnston syndrome and G6PD for rasburicase-associated hemolytic anemia; 12 (40%) as recommended, including CYP2D6 for codeine toxicity; and 14 (47%) as not routinely recommended, such as CYP2C19 for clopidogrel dosing.
Conclusion
Only half of drug–gene pairs with high clinical validity received Subcommittee support for policy promoting their widespread use across VHA. The Subcommittee generally found insufficient evidence of clinical utility or available, effective alternative strategies for the remainders. Continual evidence review and rigorous outcomes research will help promote the translation of pharmacogenetic discovery to healthcare.
Similar content being viewed by others


Introduction
Defined as the study of how genotype impacts the efficacy and toxicity an individual experiences from medications, pharmacogenetics has been touted as an early clinical application of genomics that might have a transformative impact on the routine practice of medicine.1,2,3 The US Food and Drug Administration (FDA) labels for almost 200 medications include pharmacogenetics considerations,4 and a recent analysis found that 91% of approximately 10,000 patients in one large healthcare system had at least one “actionable” pharmacogenetic variant associated with one of five commonly used medications.5 Catalyzed in part by advances in genome sequencing technology and the increasing availability of large patient data sets, pharmacogenetic discovery has seen exponential growth in recent years.6, 7 Many healthcare systems are now integrating large pharmacogenetics programs into their patient care activities, through research studies, clinical implementation projects, or a combination of both.8,9,10,11,12,13,14
The Veterans Health Administration (VHA) of the US Department of Veterans Affairs is the largest integrated healthcare system in the United States, providing primary and specialty care for more than 9 million veterans each year, spanning every state and US territory. Policymaking and oversight of laboratory testing in the VHA occurs at several levels, from the Office of the National Director of the Pathology and Laboratory Medicine Service (P&LMS) to the more than 145 P&LMS Chiefs or Directors and Administrative Laboratory Chiefs at local and regional VHA facilities nationwide.15 The P&LMS Chief or Director at each VHA facility is the steward of that facility’s P&LMS budget and is responsible for overseeing all in-house and ancillary laboratory testing ordered at that facility, including the evaluation of a given test’s appropriateness.
However, the VHA currently lacks a standardized national approach to the clinical use of pharmacogenetic testing. Despite a substantial body of literature supporting the clinical validity of several pharmacogenetic associations, there has been a paucity of evidence of clinical utility and cost-effectiveness with respect to many of the pharmacogenetic tests in variable use across VHA locations. Clinicians might overutilize certain tests based on vendors’ recommendations, political pressure, or practices from academic affiliations. On the other hand, they might underutilize tests due to lack of knowledge or systems-level barriers to test ordering. With its common electronic health record (EHR) that includes both laboratory test results and pharmacy records, VHA is uniquely positioned to use and benefit from pharmacogenetic testing in patient care. However, the variation in clinical pharmacogenetics practice across the health system contributes to a lack of consensus on whether the costs of pharmacogenetic testing should be borne by the P&LMS, Pharmacy Service, or both at VHA locations.
In 2013, the National Director of P&LMS created a Molecular Diagnostics Working Group to develop a standardized VHA approach to molecular genetics diagnostic testing. The aims of the working group were to provide recommendations on how to effectively utilize molecular genetic tests, promote the quality and availability of testing across VHA, encourage internal referral testing, and inform P&LMS policy for molecular genetic testing and laboratory developed tests. The VHA Molecular Diagnostics Working Group consists of several subcommittees, including Oncology, Germline/Inherited Genetics, Hematopathology, Microbial Genetics, and Clinical Pharmacogenetics. To illustrate how one large integrated health system is approaching policymaking around pharmacogenetic testing, here we describe the first two years of the Clinical Pharmacogenetics Subcommittee’s activities in reviewing the scientific evidence for and making policy recommendations about its routine use.
Materials and methods
Clinical Pharmacogenetics Subcommittee charge and consensus definitions
The Clinical Pharmacogenetics Subcommittee (hereafter, the “Subcommittee”), is comprised of VHA and non-VHA experts in the fields of pharmacology, laboratory medicine, medical genetics, and health services research. In 2014, the Molecular Diagnostics Working Group recognized the lack of consistency in how VHA laboratories across the United States were using pharmacogenetic testing in clinical care. For example, there was variable use of CYP2D6 and CYP2C19 genotyping to guide pharmacotherapy for major depressive disorder. To address this lack of policy and uniform practice, the Molecular Diagnostics Working Group charged the Subcommittee with making recommendations for standardizing pharmacogenetic testing across VHA. These recommendations would then be forwarded to the VHA Office of Specialty Care Services to inform VHA policymaking around pharmacogenetic testing.
When the Subcommittee first convened, it recognized the need for a process to evaluate specific pharmacogenetic tests. Members understood that the strength of the scientific evidence supporting individual drug–gene and drug–variant associations is variable,16, 17 and they were familiar with several existing resources and professional organizations endeavoring to standardize the field. These included PharmGKB (https://www.pharmgkb.org), sponsored by the US National Institutes of Health, which curates knowledge about the impact of genetic variation on drug response for clinicians and researchers.18 Some Subcommittee members were also members of the Clinical Pharmacogenetics Implementation Consortium (CPIC, https://cpicpgx.org), which performs systematic evidence grading of pharmacogenetic tests and, going one step further in clinical translation than PharmGKB, issues prescribing and dosing recommendations for specific medications if genotype information is available.17, 19
The Subcommittee recognized that its role was not to recreate the work of these organizations but, rather, to incorporate and adapt it to inform policy recommendations for whether and how to use pharmacogenetic testing in VHA patient care. This recognition led to a discussion of the distinction between clinical validity and clinical utility, informed largely by the hierarchical ACCE model that evaluates a genetic test by its Analytic validity, Clinical validity, Clinical utility, and Ethical, legal, and social implications.20 To guide its activities, the Subcommittee used a consensus approach to generate the following working definitions of these key concepts specific to pharmacogenetic testing:
Pharmacogenetics is the study of inherited genetic differences in drug metabolic pathways (absorption, distribution, metabolism, or excretion) which can affect individual responses to drugs in terms of therapeutic and/or adverse effects. The analytic validity of a pharmacogenetic test refers to the ability of the test to accurately identify the genotype of interest. The clinical validity of a pharmacogenetic test result refers to the likelihood that the test result (genotype) provides information about therapeutic efficacy or toxicity. The clinical utility of a pharmacogenetic test result refers to the likelihood that pharmacogenetic information will lead to a change in clinical management that improves health outcomes. The clinical utility of pharmacogenetics also describes its ability to identify the right drug or drug combinations at the correct dosages, thereby maximizing benefits to the patient while minimizing toxicity.
Using these definitions, the Subcommittee recognized that the work of PharmGKB and CPIC most closely aligned with the concepts of analytic and clinical validity and that CPIC dosing guidelines were designed to help clinicians understand how available pharmacogenetic test results should be used to optimize drug therapy, rather than whether tests should be ordered. The Subcommittee thus specified its aim to evaluate the evidence supporting the clinical utility of specific pharmacogenetic tests, using the above definitions.
Choice of drug–gene pairs to review
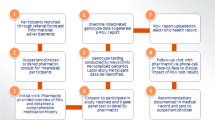
To generate a list of pharmacogenetic tests with potential clinical utility, the Subcommittee identified drug–gene pairs from PharmGKB and CPIC with clinically valid genotype–phenotype associations, drawing predominantly from CPIC Level A, which indicates a strong recommendation that specific pharmacogenetic results, if known, should be used to inform the prescribing of the drug in question.17 The Subcommittee excluded drug–gene pairs addressed by other Molecular Diagnostics Working Group subcommittees, including FDA-approved companion diagnostics for oncology targeted therapy such as afatinib-EGFR, crizotinib-ALK, erlotinib-EGFR, and trametinib-BRAF V600. Drug–gene pairs relevant exclusively to pediatric populations were not considered. By March 2015, the Subcommittee had chosen 30 drug–gene pairs for review (Table 1).
Evidence review and synopsis
Subcommittee members were each assigned drug–gene pairs to review. For each drug–gene pair, the reviewer was asked to complete a review template summarizing the indications for drug use, mechanism of action, pharmacokinetics, and pharmacogenetics of the medication (Supplemental Materials and Methods). Reviewers consulted the databases of clinical and research pharmacogenetic groups from the government and private sector, including the FDA, the PharmacoGenomics Mutation Database (PGMD), the Human Cytochrome P450 Allele Nomenclature Database (now a part of the PharmVar Consortium21), PharmaADME,22 the International Serious Adverse Events Consortium (iSAEC),23 and the UCSF Pharmacogenetics of Membrane Transporters (PMT) Database.24 Each reviewer was also asked to include PharmGKB and CPIC levels of evidence, in addition to any pharmacogenetic considerations on the drug’s FDA label. As applicable, each reviewer listed specific gene variants included in their review.
Consensus process for pharmacogenetic testing recommendations
On monthly teleconferences, members presented their review summaries to the Subcommittee. All members had the opportunity to ask questions and discuss the clinical relevance and utility of testing for that drug–gene association in clinical care across the VHA. Based on the review summary and the ensuing discussion, the presenting Subcommittee member was asked to provide a recommendation for whether routine pharmacogenetic testing for that drug–gene association should be (1) strongly recommended for the use of that drug in VHA, (2) recommended for the use of that drug in VHA, or (3) not routinely recommended for the use of that drug in VHA. Of note, the Subcommittee intended these recommendations for general application to the routine initiation of the medications in question, and members had the option to add annotations for specific clinical contexts and patient characteristics for which the recommendations might differ from the general recommendation. The Subcommittee did not explicitly consider genotype frequency or the costs of testing in its deliberation. If a quorum was present, the Subcommittee voted by majority rule on whether to approve the recommendation. It is important to note that the Subcommittee’s evidence review and ensuing recommendations did not establish VHA policy but, rather, would be forwarded as policy guidance to the VHA Office of Specialty Care Services.
Results
Subcommittee recommendations
The Subcommittee reviewed 30 drug–gene pairs between February 2016 and April 2017 (Table 1 and Supplemental Materials and Methods). By design, all 30 had CPIC recommendations of Level A strength. All but two had PharmGKB level of evidence 1A at the time of review (Table 1), indicating variant–drug combinations with strong evidence of genotype–phenotype association (clinical validity) that have a CPIC or other medical society–endorsed pharmacogenetic guideline or are implemented at a Pharmacogenomics Research Network site or in another major health system.18
The Subcommittee determined that 4 (13%) of the 30 pharmacogenetic tests should be strongly recommended before prescribing the associated drug, 12 (40%) should be recommended, and 14 (47%) should not be routinely recommended for clinical care across the VHA (Table 1 and Supplemental Materials and Methods). Table 1 summarizes the Subcommittee’s deliberations about each drug–gene pair. Although this rubric was not defined a priori, the Subcommittee’s recommendations point to some general observations. Testing tended to be strongly recommended if the phenotype was a severe adverse drug effect (ADE) that could be avoided with alternative therapy. Recommended tests could inform either the risk of ADE or drug efficacy; tests related to an ADE tended to be recommended and not strongly recommended if the associated medication was already in very widespread use across VHA (e.g., allopurinol and phenytoin), potentially making system-wide implementation unwieldy. Tests that were not routinely recommended tended to inform drug efficacy but lacked studies demonstrating improved patient outcomes. The following examples illustrate these three categories and the Subcommittee’s associated discussions.
Strongly recommended: HLA-B*57:01 genotyping for abacavir use
Abacavir is a nucleoside analog reverse transcriptase inhibitor used in the treatment of human immunodeficiency virus (HIV) infection. The medication is generally well tolerated, but 5–8% of patients experience a hypersensitivity reaction during the first 6 weeks of treatment, characterized by rash, fever, fatigue, cough, gastrointestinal symptoms, hypotension, and possibly death. The association between human leukocyte antigen (HLA)-B*57:01 and abacavir hypersensitivity has been replicated in several studies, and a large prospective randomized controlled trial has demonstrated the complete avoidance of hypersensitivity reactions among patients prescreened for HLA-B*57:01 prior to therapy. Since 2008, the FDA label for abacavir has recommended HLA-B*57:01 testing prior to initiation and has issued a boxed warning that abacavir not be prescribed to any patient testing positive for HLA-B*57:01.25
The Subcommittee endorsed strongly recommending HLA-B*57:01 genotyping prior to abacavir use in the VHA. Factors supporting this recommendation included the availability of strong evidence for the clinical validity of the association between HLA-B*57:01 genotype and abacavir hypersensitivity and the high negative predictive value of a negative HLA-B*57:01 genetic result. Perhaps more importantly, the Subcommittee valued the availability of randomized trial data demonstrating improved patient outcomes (i.e., avoidance of hypersensitivity reactions after a prescreening intervention) and the ability of HLA-B*57:01 results to inform the choice between therapy with abacavir and other available antiretroviral medications.
Recommended: CYP2D6 genotyping for codeine use
Codeine is an opioid agonist analgesic with relative selectivity for the μ-opioid receptor. It is metabolized to morphine in the liver by cytochrome P450 2D6 (CYP2D6). As a result, high codeine intake can cause morphine intoxication, characterized by sedation, confusion, respiratory depression, and even death. The CYP2D6 gene is highly polymorphic, but genotypes can be broadly classified into poor, intermediate, normal (formerly referred to as extensive), and ultrarapid metabolizers. In its 2012 and 2014 guidelines, CPIC recommended that codeine not be prescribed for patients with ultrarapid metabolizer genotypes, given the risk of toxicity. At the other extreme, CPIC also recommended that codeine be avoided for poor metabolizers, given the possible analgesic inefficacy of codeine in those individuals.26
The Subcommittee recommended that prescribers consider CYP2D6 genotyping prior to codeine use in the VHA. Factors supporting this recommendation again included the actionability of the genetic test result, guiding prescribers to different codeine dosing or alternate analgesic agents. Members acknowledged the pressing concerns of the current opioid use disorder epidemic across the United States broadly and the VHA specifically and valued the potential of this test to guide more thoughtful approaches to pain management. The Subcommittee did not vote to strongly recommend CYP2D6 genotyping prior to codeine use, however. They reasoned that codeine is rarely prescribed as a single agent or for long-term use in the VHA patient population, more commonly prescribed in cough syrups (e.g., guaifenesin–codeine) or in combination with acetaminophen (Tylenol® No. 3).
Not routinely recommended: CYP2C19 genotyping for clopidogrel use
Clopidogrel is a widely used prodrug that inhibits platelet aggregation, used in the treatment of cardiovascular diseases including acute coronary syndrome, stroke, and peripheral arterial disease. It is metabolized to its active metabolite predominantly by CYP2C19, whose haplotypes can be categorized as poor, intermediate, normal, rapid, and ultrarapid metabolizers. The 2013 CPIC guidelines recommend that patients who are intermediate or poor metabolizers based on CYP2C19 genotype be prescribed alternate antiplatelet therapy, such as prasugrel or ticagrelor, out of concern for lack of efficacy and the possibility of increased incidence of cardiovascular events.27
The Subcommittee did not routinely recommend CYP2C19 genotyping for the initiation of clopidogrel in VHA. Members considered a few factors in arriving at this recommendation. First, the Subcommittee acknowledged the availability of an alternate test, such as platelet aggregometry, that could produce similar clinical information. Second, clopidogrel is widely used across VHA, and a recommendation for routine CYPC19 testing could place tremendous burden on the healthcare system, given the dearth of VHA laboratories able to perform CYPC19 genotyping. Third, no randomized trial had demonstrated improved clinical outcomes from CYP2C19 testing, such as lower cardiovascular event rates. Fourth, American Heart Association guidelines did not recommend the routine use of CYP2C19 testing in acute coronary syndrome. However, in its recommendation, the Subcommittee did acknowledge specific clinical contexts in which CYP2C19 genotyping might be considered, such as patients who have had a cardiovascular event while on clopidogrel or patients who are at high risk for poor outcomes (Table 1).
Discussion
In its first two years, the VHA Clinical Pharmacogenetics Subcommittee of the Molecular Diagnostics Working Group has made policy recommendations regarding the routine use of 30 pharmacogenetic tests in clinical care at VHA facilities. Despite high grades of PharmGKB evidence supporting these drug–gene associations and strong CPIC recommendations for drug dosing if associated genotype is known, the Subcommittee voted to strongly recommend or recommend only about half of the tests it reviewed. Considered with other factors such as feasibility, cost, and patient and provider acceptance, these recommendations will inform national VHA policy for pharmacogenetic testing.
The Subcommittee’s recommendations do not contradict the work of PharmGKB or CPIC but, rather, highlight the need for demonstrated improvements in patient outcomes before large healthcare systems might broadly implement pharmacogenetic testing outside of a research context. PharmGKB evaluates the clinical validity of drug–gene pairs, that is, the evidence supporting the association of a given gene or variant with a drug phenotype. CPIC issues guidelines for drug dosing when genotype is already known but does not make recommendations on whether testing should be initiated in the first place. The gap between the work of PharmGKB and CPIC and the Subcommittee’s recommendations is one of clinical utility, a reason often cited by payers for not covering new laboratory tests.7, 28, 29 There is no universally accepted definition of clinical utility.30, 31 However, for the pharmacogenetics context, the Subcommittee’s consensus definition (“likelihood that pharmacogenetic information will lead to a change in clinical management that improves health outcomes”) refers to the impact of testing on at least two related categories of outcomes. First, clinical utility encompasses actionability, evidence that the pharmacogenetic result changes healthcare providers’ clinical management in terms of drug choice and dose.32, 33 Management change would seem to be the minimum necessary demonstration of a pharmacogenetic test’s clinical utility, consistent with the conventional clinical wisdom of not ordering a test unless its results will change medical decision-making. Second, a pharmacogenetic test ideally has prospectively collected evidence that the management change it prompts results in improved clinical outcomes, such as lower rates of adverse drug effects (drug safety) or prevention or better treatment of the associated condition (drug efficacy). The Subcommittee’s deliberations underscored the dearth of such evidence for many drug–gene pairs, despite their high clinical validity.
Other healthcare delivery systems have grappled with the question of which pharmacogenetic tests to implement in their clinical activities, using varying criteria to guide decision-making. The National Institutes of Health Clinical Center chose HLA genotyping to launch its Pharmacogenetics Testing Implementation Committee because of local capabilities.34 St. Jude Children’s Research Hospital launched its pharmacist-managed Clinical Pharmacogenetics Service with thiopurine methyltransferase (TPMT) and uridine glucuronosyltransferase 1A1 (UGT1A1) testing, guided not only by the importance of azathioprine, mercaptopurine, thioguanine, and irinotecan in pediatric oncology but also by Phillips’ recommendations that a clinically important pharmacogenetic test applies to a commonly used medication with a high incidence of severe toxicity and a relatively common at-risk genotype, among other criteria.35, 36 Institutions where patient genotypes are already preemptively available through clinical care, biobanks, or other research projects often use CPIC recommendations to choose which pharmacogenetic results to implement clinically.37 At the national level, the Centers for Disease Control and Prevention (CDC) Office of Public Health Genomics performs horizon scanning of FDA policies, Centers for Medicare & Medicaid Services coverage decisions, clinical practice guidelines, and systematic reviews to rank the evidence supporting the clinical use of genomic tests, including certain pharmacogenetic tests.38 All four of the Subcommittee’s strongly recommended drug–gene pairs are categorized as “Tier 1” by the CDC, indicating readiness for clinical implementation.38, 39 No one approach to evidence review and policymaking will apply to all healthcare contexts.
It is likely that some external pharmacogenetics experts and stakeholders will disagree with the Subcommittee’s recommendations for specific drug–gene pairs. We do not intend for these recommendations to supplant the work of organizations such as PharmGKB and CPIC. Instead, we present them to illustrate how one large integrated health system is building on their work and using context-specific considerations to inform policymaking around pharmacogenetic testing. It is important to note that the Subcommittee’s recommendations are meant to serve as guidance, not dogma, for VHA policymakers. The Subcommittee initially considered designating some drug–gene pairs as required, modeled after FDA black box warnings, but members expressed unease using this term, reluctant to apply a rigid designation suggesting that not testing would be inappropriate care, particularly in the absence of systems to facilitate testing. Within the Subcommittee, members held differing opinions on whether evidence from large randomized controlled trials (RCTs) is necessary to determine clinical utility, recognizing the impossibility of conducting RCTs to study all potential subgroups of patients who might benefit from pharmacogenetic testing. The Subcommittee also intends to update its recommendations and annotations as new scientific evidence accrues. For example, results of an RCT published after April 2017 prompted the Subcommittee to add an annotation that pharmacogenetic testing might be recommended for patients initiating perioperative warfarin for elective orthopedic surgery.40
This work has limitations to note. The Subcommittee used targeted evidence review of resources including PharmGKB and CPIC and did not perform independent systematic literature reviews to guide its deliberations. Second, its recommendations apply only to reactive pharmacogenetic testing, or test ordering at the clinical moment of medical decision-making around pharmacotherapy. Many have argued that preemptive testing, whereby patients undergo pharmacogenetic (or even broader genomic) testing early in the life course to inform medical care, will prove a more efficient and even cost-effective model;8, 9 indeed, CPIC guidelines rest on this premise17 and thus cannot fully inform the Subcommittee’s work. Third, the Subcommittee’s recommendations apply only to testing for individual drug–gene pairs instead of pharmacogenetic panel testing, which may provide efficiencies of scale as the costs of genotyping decrease. Fourth, the Subcommittee’s specific recommendations cannot necessarily be generalized to healthcare settings outside VHA; still, this description of the process of VHA policy recommendation may be informative for other health systems. Fifth, the Subcommittee did not explicitly consider the costs of pharmacogenetic testing in its deliberation; any VHA policies stemming from the Subcommittee’s recommendations will need to do so.
As the VHA Office of Specialty Care Services considers incorporating the Subcommittee’s recommendations into VHA policy, implementation and dissemination strategies will be needed to promote appropriate testing in the healthcare system. One strategy to decrease inappropriate pharmacogenetic testing is already in place in the VA San Diego Healthcare System (VASDHS), where a healthcare provider must request a pharmacogenomics e-consult through the EHR to obtain approval from a clinical pharmacogenetic expert before ordering certain tests. This pharmacist reviews the literature and identifies the strength of the evidence linking the test to the medication to be prescribed, evaluates the benefit of the test to the medication, and approves or disapproves the test. Once approved, the VASDHS clinical laboratory processes the test for send-out. The pharmacist expert writes a consult note to inform the providers about the proper utilization, risks, benefits, and costs of using the test results for the medication. Strategies to increase appropriate pharmacogenetic testing also include academic detailing and pharmacist review of prescriptions for medications requiring pharmacogenetic testing. Additionally, appropriate testing can be promoted through clinical decision support integrated into the EHR, including web-based point-of-care resources to educate clinicians and alerts prompting prescribers to consider testing at the clinical moment a medication is prescribed.34
The Subcommittee’s recommendations and annotations will be continually updated as new evidence accrues. To accelerate such evidence generation, the VHA Office of Research & Development is currently funding ongoing RCTs of pharmacogenetic testing in the treatment of major depressive disorder (ClinicalTrials.gov Identifier: NCT03170362) and cardiovascular risk reduction (ClinicalTrials.gov Identifier: NCT02871934). The Subcommittee’s work highlights the need for additional rigorous outcomes research to promote the translation of pharmacogenetic discovery into patient care.
References
Manolio TA, Chisholm RL, Ozenberger B, et al. Implementing genomic medicine in the clinic: the future is here. Genet Med . 2013;15:258–67.
Relling MV, Evans WE. Pharmacogenomics in the clinic. Nature. 2015;526:343–50.
Drew L. Pharmacogenetics: the right drug for you. Nature. 2016;537:S60–62.
United States Food and Drug Administration. Table of pharmacogenomic biomarkers in drug labels. 2017. https://www.fda.gov/Drugs/ScienceResearch/ucm572698.htm. Accessed 24 March 2018.
Van Driest SL, Shi Y, Bowton EA, et al. Clinically actionable genotypes among 10,000 patients with preemptive pharmacogenomic testing. Clin Pharmacol Ther. 2014;95:423–31.
Scott SA. Personalizing medicine with clinical pharmacogenetics. Genet Med. 2011;13:987–95.
Patel HN, Ursan ID, Zueger PM, et al. Stakeholder views on pharmacogenomic testing. Pharmacotherapy. 2014;34:151–65.
Dunnenberger HM, Crews KR, Hoffman JM, et al. Preemptive clinical pharmacogenetics implementation: current programs in five US medical centers. Annu Rev Pharmacol Toxicol. 2015;55:89–106.
Bielinski SJ, Olson JE, Pathak J, et al. Preemptive genotyping for personalized medicine: design of the right drug, right dose, right time-using genomic data to individualize treatment protocol. Mayo Clin Proc. 2014;89:25–33.
Rasmussen-Torvik LJ, Stallings SC, Gordon AS, et al. Design and anticipated outcomes of the eMERGE-PGx project: a multicenter pilot for preemptive pharmacogenomics in electronic health record systems. Clin Pharmacol Ther. 2014;96:482–9.
Luzum JA, Pakyz RE, Elsey AR, et al. The Pharmacogenomics Research Network Translational Pharmacogenetics Program: outcomes and metrics of pharmacogenetic implementations across diverse healthcare systems. Clin Pharmacol Ther. 2017;102:502–10.
O’Donnell PH, Danahey K, Jacobs M, et al. Adoption of a clinical pharmacogenomics implementation program during outpatient care--initial results of the University of Chicago “1200 Patients Project”. Am J Med Genet C Semin Med Genet . 2014;166c:68–75.
van der Wouden CH, Cambon-Thomsen A, Cecchin E, et al. Implementing pharmacogenomics in Europe: design and implementation strategy of the Ubiquitous Pharmacogenomics Consortium. Clin Pharmacol Ther. 2017;101:341–58.
Empey PE, Stevenson JM, Tuteja S, et al. Multi-site investigation of strategies for the implementation of CYP2C19 genotype-guided antiplatelet therapy. Clin Pharmacol Ther. 2017. Dec 26. https://doi.org/10.1002/cpt.1006. [Epub ahead of print].
Department of Veterans Affairs. VHA handbook 1106.01: pathology and laboratory medicine service procedures. 2016. https://www.va.gov/vhapublications/ViewPublication.asp?pub_ID=3169. Accessed 24 March 2018.
Wang B, Canestaro WJ, Choudhry NK. Clinical evidence supporting pharmacogenomic biomarker testing provided in US Food and Drug Administration drug labels. JAMA Intern Med. 2014;174:1938–44.
Caudle KE, Klein TE, Hoffman JM, et al. Incorporation of pharmacogenomics into routine clinical practice: the Clinical Pharmacogenetics Implementation Consortium (CPIC) guideline development process. Curr Drug Metab. 2014;15:209–17.
Whirl-Carrillo M, McDonagh EM, Hebert JM, et al. Pharmacogenomics knowledge for personalized medicine. Clin Pharmacol Ther . 2012;92:414–7.
Relling MV, Klein TE. CPIC: Clinical Pharmacogenetics Implementation Consortium of the Pharmacogenomics Research Network. Clin Pharmacol Ther. 2011;89:464–7.
Haddow J, Palomaki G. ACCE: a model process for evaluating data on emerging genetic tests. In: Khoury M, Little J, Burke W, editors. Human genome epidemiology. Oxford: Oxford University Press; 2004. p. 217–33.
Gaedigk A, Ingelman-Sundberg M, Miller NA, et al. The Pharmacogene Variation (PharmVar) Consortium: incorporation of the Human Cytochrome P450 (CYP) Allele Nomenclature Database. Clin Pharmacol Ther. 2017;103:399–401.
PharmaADME. 2017. http://www.pharmaadme.org/. Accessed 24 March 2018.
International SAE Consortium. 2017. https://www.saeconsortium.org/. Accessed 24 March 2018.
Pharmacogenetics of Membrane Transporters Database. 2015. http://pharmacogenetics.ucsf.edu/. Accessed 24 March 2018.
Martin MA, Klein TE, Dong BJ, et al. Clinical Pharmacogenetics Implementation Consortium guidelines for HLA-B genotype and abacavir dosing. Clin Pharmacol Ther. 2012;91:734–8.
Crews KR, Gaedigk A, Dunnenberger HM, et al. Clinical Pharmacogenetics Implementation Consortium guidelines for cytochrome P450 2D6 genotype and codeine therapy: 2014 update. Clin Pharmacol Ther. 2014;95:376–82.
Scott SA, Sangkuhl K, Stein CM, et al. Clinical Pharmacogenetics Implementation Consortium guidelines for CYP2C19 genotype and clopidogrel therapy: 2013 update. Clin Pharmacol Ther. 2013;94:317–23.
Keeling NJ, Rosenthal MM, West-Strum D, et al. Preemptive pharmacogenetic testing: exploring the knowledge and perspectives of US payers. Genet Med. 2017. Oct 26. https://doi.org/10.1038/gim.2017.181. [Epub ahead of print].
Hess GP, Fonseca E, Scott R, et al. Pharmacogenomic and pharmacogenetic-guided therapy as a tool in precision medicine: current state and factors impacting acceptance by stakeholders. Genet Res. 2015;97:e13.
ACMG Board of Directors. Clinical utility of genetic and genomic services: a position statement of the American College of Medical Genetics and Genomics. Genet Med. 2015;17:505.
Drozda K, Pacanowski MA. Clinical trial designs to support clinical utility of pharmacogenomic testing. Pharmacotherapy. 2017;37:1000–4.
Snyder SR, Mitropoulou C, Patrinos GP, et al. Economic evaluation of pharmacogenomics: a value-based approach to pragmatic decision making in the face of complexity. Public Health Genomics. 2014;17:256–64.
O’Donnell PH, Wadhwa N, Danahey K, et al. Pharmacogenomics-based point-of-care clinical decision support significantly alters drug prescribing. Clin Pharmacol Ther. 2017;102:859–69.
Goldspiel BR, Flegel WA, DiPatrizio G, et al. Integrating pharmacogenetic information and clinical decision support into the electronic health record. J Am Med Inform Assoc. 2014;21:522–8.
Crews KR, Cross SJ, McCormick JN, et al. Development and implementation of a pharmacist-managed clinical pharmacogenetics service. Am J Health Syst Pharm. 2011;68:143–50.
Phillips KA, Veenstra DL, Oren E, et al. Potential role of pharmacogenomics in reducing adverse drug reactions: a systematic review. JAMA. 2001;286:2270–9.
Sperber NR, Carpenter JS, Cavallari LH, et al. Challenges and strategies for implementing genomic services in diverse settings: experiences from the Implementing GeNomics In pracTicE (IGNITE) network. BMC Med Genomics. 2017;10:35.
Dotson WD, Douglas MP, Kolor K, et al. Prioritizing genomic applications for action by level of evidence: a horizon-scanning method. Clin Pharmacol Ther. 2014;95:394–402.
Centers for Disease Control and Prevention Office of Public Health Genomics. Public Health Genomics Knowledge Base (v2.1): Tier TableDatabase. 2017. https://phgkb.cdc.gov/PHGKB/topicFinder.action?Mysubmit=init&query=all. Accessed 24 March 2018.
Gage BF, Bass AR, Lin H, et al. Effect of genotype-guided warfarin dosing on clinical events and anticoagulation control among patients undergoing hip or knee arthroplasty: the GIFT randomized clinical trial. JAMA. 2017;318:1115–24.
Acknowledgements
JLV is supported by Career Development Award IK2-CX001262 from the Clinical Sciences Research and Development (CSR&D) Service of the US Department of Veterans Affairs (VA). VMP is supported by the IGNITE project grant (U01-HG007762) from the National Institutes of Health and the Indiana University Health–Indiana University School of Medicine Strategic Research Initiative. The Pharmacogenomics Analysis Laboratory at the Central Arkansas Veterans Healthcare System is funded by the Cooperative Studies Program of the VA CSR&D Service. The contents do not necessarily represent the views of the US government or the VA.
Author information
Authors and Affiliations
Consortia
Corresponding author
Ethics declarations
DISCLOSURE
The authors declare no conflicts of interest.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Vassy, J.L., Stone, A., Callaghan, J.T. et al. Pharmacogenetic testing in the Veterans Health Administration (VHA): policy recommendations from the VHA Clinical Pharmacogenetics Subcommittee. Genet Med 21, 382–390 (2019). https://doi.org/10.1038/s41436-018-0057-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-018-0057-x
Keywords
This article is cited by
-
Patients dispensed medications with actionable pharmacogenomic biomarkers: rates and characteristics
Genetics in Medicine (2021)
-
Prevalence and types of inconsistencies in clinical pharmacogenetic recommendations among major U.S. sources
npj Genomic Medicine (2020)
-
Considerations for pharmacogenomic testing in a health system
Genetics in Medicine (2019)
