
Abstract
Purpose
Quantitative definition of the natural history of free sialic acid storage disease (SASD, OMIM 604369), an orphan disorder due to the deficiency of the proton-driven carrier SLC17A5.
Methods
Analysis of published cases with SASD (N = 116) respecting STROBE criteria. Main outcome parameters: survival and diagnostic delay. Phenotype, phenotype–biomarker associations, and geographical patient distribution were explored.
Results
Median age at disease onset was 0.17 years. Median age at diagnosis was 3 years with a median diagnostic delay of 2.5 years. Median survival was 11 years. The biochemical phenotype clearly predicted the disease course: patients with a urinary free sialic acid excretion below 6.37-fold or an intracellular free sialic acid storage in fibroblasts below 7.37-fold of the mean of normal survived longer than patients with biochemical values above these thresholds. Cluster analysis of disease features suggested a continuous phenotypic spectrum. Patient distribution was panethnic.
Conclusion
Combination of neurologic symptoms, visceromegaly, and dysmorphic features and/or nonimmune hydrops fetalis should prompt specific tests for SASD, reducing diagnostic delay. The present quantitative data inform clinical studies and may stimulate and accelerate development of specific therapies. Biomarker–phenotype association is particularly important for both counseling parents and study design.
Similar content being viewed by others

Introduction
Free sialic acid storage disease (SASD; OMIM 604369) is a rare progressive autosomal recessive neurodegenerative disorder manifesting mainly with muscular hypotonia, cerebellar ataxia, and cognitive impairment, as well as visceromegaly and coarse facial features.1,2 The disease is caused by biallelic pathogenic variants in the SLC17A5 gene (cytogenetic location 6q13) encoding a proton-driven carrier for sialic acid across lysosomal membranes.1,3 Salla disease, the Finnish form of SASD, was first described by Aula in 1979 (ref. 4) and infantile free sialic acid storage disease was described in 1982 by Tondeur.5 Lysosomes in patients are enlarged at electron microscopic examination; in addition, abnormally high levels of free sialic acid are excreted in urine.1 Intriguingly, SLC17A5 also acts as vesicular glutamic acid–aspartate cotransporter in the brain and impaired energy-dependent aspartergic and glutamatergic neurotransmission has been proposed as underlying cause for the observed severe central nervous system (CNS) manifestation in SASD.6 In addition, sialin-/- mice exhibit a progressive leukoencephalopathy, characterized by decreased number of myelinated axons, postmitotic oligodendrocytes associated with increased percentage of apoptotic cells during later stages of mylogenesis, ultimately leading to coordination defects, seizures, and premature death.7,8
By 31 December 2017, 36 pathognomonic variants were listed in the Human Gene Mutation Database at the University of Cardiff (http://www.hgmd.cf.ac.uk/ac/gene.php?gene=SLC17A5, accessed 31 December 2017). Precise prevalence and incidence are unknown; Orphanet lists 130 known cases.9 Currently, there are no clinical trials in humans listed on clinicaltrials.gov (accessed 31 December 2017). No orphan drug designation was granted for any compound intended to treat SASD.10 Quantitative data especially regarding survival rates of patients with SASD are not available so far. Information on survival is important for counseling parents of afflicted children. Moreover, to assess the long-term impact of potential novel therapeutic approaches reliable data on the survival of untreated patients as hard clinical endpoint for future interventional trials would be indispensable. In addition, SASD is an ultrarare metabolic disorder that renders diagnostic suspicion difficult. Because establishing the diagnosis SASD requires specialized assays that are not available in a routine care setting or even in many countries, the risk of a considerable diagnostic delay is high. For SASD the precise diagnostic delay has not been defined. However, as soon as novel therapies are developed, it would be important to further raise disease awareness to accelerate the diagnostic process, taking into account the concept of a limited window of opportunity for early intervention, which might be associated with a beneficial outcome for afflicted individuals. Given the rarity of the condition, conducting a global prospective natural history study would be virtually unfeasible. Important information, especially on hard clinical endpoints, can be obtained from systematic analyses of published cases and case series in the literature in a timely and efficient manner.11,12,13 Therefore, the purpose of the present study is to quantitatively analyze the onset of disease, diagnostic delay, clinical characteristics, and survival rates of the published evidence on SASD. Because disease onset and survival rates are often a function of the residual enzymatic activities and/or the amount of macromolecules excreted or stored intracellularly in lysosomal storage disorders,12,13 we also test the hypothesis of biomarker–phenotype association for SASD. In addition, we analyze the geographical distribution of patients worldwide, which might be of interest for epidemiological reasons or the planning of future prospective clinical trials.
Materials and methods
This analysis was conducted according to STROBE (STrengthening the Reporting of OBservational studies in Epidemiology) criteria (http://www.strobe-statement.org).14
Literature review and definitions of variables
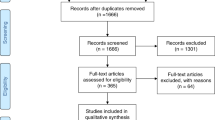
We conducted a comprehensive literature search on PubMed using the keywords “sialic acid storage disease,” “Salla disease,” and “SLC17A5 deficiency.” Identified publications were downloaded (N = 252) and manually sorted for reports containing relevant clinical, biochemical, and/or genetic information (N = 89). Selected reports were published between 1976 and 2015. In total, we identified 116 patients from 74 publications with sufficient information for analysis (Supplementary Fig. 1). The articles were published in English, German, or French. Close of database was 30 June 2017. None of the analyzed patients underwent bone marrow transplantation.
The following variables were extracted: age at onset, age at diagnosis, symptoms leading to diagnosis, last reported age, information on whether the patient is alive or deceased, urinary excretion of free and total sialic acid, free or total sialic acid content in serum, amniotic fluid and cerebrospinal fluid, intracellular storage of free and total sialic acid in fibroblasts or leukocytes, publication year, and origin of patients. If the origin of the patient has not been explicitly stated, the country of patient’s origin has been attributed to the country of the first author’s institutional affiliation in the respective case description.
If information regarding time was stated in semiquantitive terms in the respective publications, we took a conservative approach and defined the findings as follows: “stillborn” = day 0, “newborn” or “at birth” = day 1, “newborn period “ = 1 month, “post mortem” = age at death.
Statistical analysis
Techniques of descriptive statistics were applied as previously reported.12,13 Variables were summarized using counts and percentages of the total study population. Because biochemical parameters in varying specimens were measured in different laboratories, values were expressed as percentage of the mean of the reported reference range. Survival was defined as the time difference between patient birth and age at death. If the patient was reported to be alive at the time of the last follow-up visit, patient data were censored. Survival was estimated with the Kaplan–Meier method. The log-rank test was applied to compare potential differences between subgroups. We used unbiased recursive partitioning to determine cut-off values for the impact of sialic acid excretion or storage and year of publication on survival in subgroup analyses.15 Diagnostic delay was calculated as difference between age at diagnosis and age of onset of disease. To assess whether there exist distinct disease subtypes in SASD or whether there is rather a continuous spectrum of phenotype, we performed a cluster analysis of reported signs and symptoms using the Ward algorithm. The results of the cluster analysis were visualized with a dendrogram.16 To evaluate the relationship between age of onset and the level of free sialic acid in urine and fibroblasts, a generalized additive regression model (GAM) with automated smoothing selections was used. In GAM, the linear relationship between the response variable and predictors are replaced by nonlinear smooth functions. The R package “mgcv” was used to fit GAM regressions. The world map was plotted using the R extension “ggmap.”17
Missing data were not imputed. Sensitivity analyses were not conducted. All analyses were performed using R (http://www.r-project.org). P values reported were two-sided. P ≤ 0.05 was considered statistically significant.
Results
We identified 116 patients from 94 families (published between 1976 and 2015) for further statistical analysis. Within the study sample, 106 patients were diagnosed postnatally due to the presentation of characteristic features, while in 10 patients the diagnosis of SASD was established prenatally. The characteristics of the study cohort are illustrated in Table 1. The origin of affected individuals is depicted in Supplementary Fig. 2. Due to missing data in specific subsets, patients had to be excluded for different subsequent data analyses, sample sizes are always indicated (N) for the corresponding analyses.
Prenatally diagnosed cohort
Ten patients were diagnosed prenatally. Of those, 8 pregnancies were terminated and 2 patients died during a later course of pregnancy or at birth. The longest survival in the prenatally diagnosed cohort was 1 day. Two patients were identified by selective testing due to a prior affected sibling. The remaining 8 patients were diagnosed because they exhibited prenatal features suggestive of SASD.
Postnatally diagnosed cohort
Descriptive statistical values are illustrated in Supplementary Table 1. In the group of postnatally diagnosed patients median age of onset of disease was 0.17 years, interquartile range (IQR) from 0 to 0.58 years (N = 90). Median age of diagnosis was 3 years, IQR from 0.77 to 8.75 years (N = 90). Median diagnostic delay was 2.50 years, IQR from 0.66 to 8.69 years (N = 90) (Fig. 1). The median estimated survival of the group of postnatally diagnosed patients (N = 106) was 11 years (Fig. 2a). Intriguingly, patients with a urinary free sialic acid excretion below 6.37-fold of the mean of the reported reference range (briefly, “normal”) survived significantly longer as opposed to patients whose urinary excretion of free sialic acid was 6.37-fold of normal or above (p < 0.001, log-rank test; N = 54; threshold determined by unbiased recursive partitioning) (Fig. 2b). Analogously, survival rates of afflicted patients inversely correlated with the amount of intracellular storage of free sialic acid in fibroblasts: patients with an intracellular free sialic acid storage below 7.37-fold of normal survived significantly longer compared with patients showing an intracellular content of free sialic acid of 7.37-fold of normal or above (p = 0.014, log-rank test; N = 46; threshold determined by unbiased recursive partitioning) (Fig. 2c). Moreover, a higher excretion of free sialic acid in urine was associated with an earlier onset of disease (F(2.57, 2.965) = 3.13, p = 0.045, GAM, N = 45) (Fig. 3a). This observation could also be corroborated for intracellular storage of free sialic acid in fibroblasts (F(4.75, 5.55) = 8.83, p < 0.001, GAM, N = 34) (Fig. 3b). In contrast, the amount of total sialic acid in all investigated specimens did not correlate with age of onset or survival.

Data were available for N = 90 patients. Horizontal lines indicate the median. The slopes of connecting lines represent the diagnostic delay between onset of the disease and the time of diagnosis. Note the logarithmic scale of the y-axis

a Estimated overall survival distribution for patients with SASD (N = 106). Censored individuals are marked with a “+“. b Estimated survival distribution for SASD patients with an urinary excretion of free sialic acid above 6.37-fold (N = 29, black line) and below or equal to 6.37-fold of normal controls (N = 25, gray line). Censored individuals are marked with a “+“. Log-rank test, p < 0.001. c Estimated survival distribution for SASD patients with intracellular storage of free sialic acid above 7.37-fold (N = 37, black line) and below or equal to 7.37-fold of normal controls (N = 9, gray line). Intracellular free sialic acid storage was measured in fibroblasts. Censored individuals are marked with a “+“. Log-rank test, p = 0.014

a Age of disease onset (in months) of postnatally diagnosed SASD patients subject to urinary excretion of free sialic acid (n-fold of normal). Each point represents a single patient (N = 45), gray line displays estimated regression curve. Generalized additive regression model, F(2.57, 2.965) = 3.13, p = 0.045. b Age of disease onset (in months) of postnatally diagnosed SASD patients subject to intracellular storage of free sialic acid (n-fold of normal) in fibroblasts. Each point represents a single patient (N = 34), gray line displays estimated regression curve. Generalized additive regression model, F(4.75, 5.55) = 8.83, p < 0.001
We analyzed the distribution of presenting or leading signs or symptoms in the study sample (Supplementary Table 2). The most common features of SASD in descending order of frequency were developmental delay or cognitive impairment, coarse facies, organomegaly, truncal muscular hypotonia, bone anomalies, facial dysmorphism, spasticity, ataxia, failure to thrive, and short stature. Hydrops fetalis as very first clinical symptom of SASD was observed in 20.7% of cases (Supplementary Table 2). Clinical feature clustering is depicted in Supplementary Figure 3. Of note, year of publication of a respective case report or case series did not affect survival rates as determined by median split.
Discussion
We quantitatively defined the natural history of the ultrarare disorder SASD in a global cohort of 116 patients, in particular hard clinical endpoints including age at disease onset, estimated survival, and seminal disease features, and delineated a biomarker–phenotype correlation; i.e., patients with a free sialic acid excretion in urine below 6.37-fold of normal or free sialic acid storage below 7.37-fold of normal in fibroblasts survived significantly longer as opposed to patients with a free sialic acid excretion or storage above these thresholds.
As in other orphan conditions,11,12,13 there was a considerable diagnostic delay: while the median onset of disease was at 0.17 years (equaling 2 months), the median time of diagnosis was 3 years, leading to a median diagnostic delay of 2.5 years. Barrier to earlier diagnosis may be (1) low disease awareness due to the fact that the condition is orphan and not necessary widely known among clinicians, (2) the fact that the disease is not treatable and may therefore not be on the priority list of a diagnostic workup that focuses primarily on treatable conditions, and (3) the necessity of access to a specialized laboratory for biochemical, and genetic workup.
Pattern recognition is important for diagnostic suspicion of SASD. The differential diagnosis of SASD should be considered once there is a clinical pattern of global developmental delay or cognitive impairment particularly affecting speech development and regression in combination with coarse facies, organomegaly, truncal muscular hypotonia, ataxia, spasticity, bone anomalies, failure to thrive, and short stature. Notably, microcephaly was reported in 15.5% of cases. Characteristic magnetic resonance image (MRI) features were brain atrophy (19.8%) with or without hypomyelination (19%) and/or thinning of corpus callosum (13.8%).
Lysosomal storage disorders are an important differential diagnosis in the clinical workup of nonimmune hydrops fetalis accounting as underlying cause for 1 to 15% of cases with the most frequent being mucopolysaccharidosis type VII, Gaucher disease, GM1-gangliosidosis, and sialidosis.18 Because every fifth SASD patient (20.7%) demonstrated hydrops fetalis as the very first clinical sign, SASD should be considered in any case of unexplained hydrops fetalis as soon as red blood alloimmunization has been ruled out.
In the case of clinical suspicion of SASD, the diagnosis should be established by measuring free sialic acid excretion in urine using thin-layer chromatography or mass spectrometry. Urinary free sialic storage excretion is usually increased from twofold up to 100-fold in afflicted individuals.19 The fluorimetric thiobarbituric acid assay is also able to detect free sialic acid in urine. However, interfering substances may lower the measurement, in particular due to chromophores, which may contribute to increased absorbance rates, ultimately causing false results.20 If elevated urinary free sialic storage excretion is detected, skin fibroblasts analysis for the determination of subcellular free sialic acid distribution might be helpful to distinguish between lysosomal storage (SASD) and cytoplasmic accumulation, the latter being the hallmark of increased sialic acid biosynthesis in sialuria, which is caused by a defective feedback inhibition due to pathogenic variants in the UDP-GlcNAc 2-epimerase (GNE)-gene.21 The diagnosis of SASD should be confirmed by Sanger sequencing of the SLC17A5 gene. If—as a “modern” diagnostic algorithm—a given patient undergoes exome sequencing for the workup of a suspected neurogenetic disorder first-line and biallelic pathogenic variants in the SLC17A5 gene are detected, subsequent biochemical confirmation of SASD is strongly recommended to confirm the functional relevance of the molecular genetic finding.
A classification into subtypes was proposed for SASD.22 Cluster analysis enabled the mathematical distinction of three statistically similar subgroups, which clinically exhibited similar generalized as well as neurological disease features. The absence of a clear-cut exclusive delineation of phenotypical features across patients in the overall study cohort supports the notion of a continuous phenotypic spectrum rather than specifically defined SASD subtypes. This observation is in line with descriptions in other lysosomal storage disorders, such as Gaucher or Farber disease.13,23
Currently, there are no clinical trials for SASD in humans listed on clinicaltrials.gov (accessed 31 December 2017). No orphan drug designation was granted for any compound intended to treat SASD.10 Drug development for SASD might be challenging owing to the rarity of the condition. Nevertheless, the identification that survival of afflicted patients clearly correlates with the amount of intracellular free sialic acid storage might be encouraging and promote future drug development. It is tempting to speculate that pharmacologically lowering intracellular sialic acid storage (e.g., by use of channel potentiators as proven beneficial for the treatment of patients with the G551D-CFTR pathogenic variant in cystic fibrosis,24,25 or antisense nucleotides targeting altered splicing of the precursor mRNA [pre-mRNA] to increase the level of normally translated protein as described for spinal muscular atrophy26,27) can be expected to have a major beneficial impact on survival as implicated by our data.
The geographical distribution pattern of reported patients suggests a panethnic pattern across all inhabited continents and includes developed and developing countries. The distribution of afflicted patients might be of special interest for the planning of future natural history and potential therapeutic studies with regard to patient recruitment.
Limitations and directions for future research
As previously described in studies applying the same methodology, quantitative natural history modeling based on case reports has some inherent important limitations, especially ascertainment bias of softer variables such as clinical features and missing data due to the absence of a standardized study protocol. Phenotype–biomarker associations in the present work rely on pooled laboratory data not analyzed in a reference laboratory according to a standardized protocol and should be corroborated in prospective studies.11,12,13,28 Supplementary Table 1 provides an indicative summary of the statistical variability of the pooled biomarker data, which may be helpful for designing and planning further prospective biomarker validation studies in SASD. The method of quantitative natural history modeling based on case reports has already been successfully applied in conditions such as molybdenum cofactor deficiency, mucopolysaccharidosis type VII (MPS VII), and Farber disease.11,12,13 However, because the precision of the applied method relies on the availability of case reports, we hope that this study will encourage reports of unpublished cases. Disease registries or protocol-driven natural history studies provide the possibility for a more thoroughly structured, protocol-guided workup; they require the availability of necessary resources in terms of time and personnel, which may not always be feasible for every single one of the plethora of orphan conditions. Supplementary Table 3 compares multiple methods to quantitatively define natural histories of orphan conditions with advantages in terms of time, potential to elaborate meaningful endpoint data, and logistic burden. The choice of method for a particular drug development program may be driven by the research question in the particular condition (e.g., organ manifestation, disease progression, nature of endpoints, and the feasibility of assessment) and may encompass a combinatory approach. Given the above limitations our approach was the most rapid and feasible method to define important outcome measures that will inform future clinical studies in ultrarare SASD. We hope that with a more profound and quantitative understanding of the natural history as provided in the present paper the development of specific therapies can be stimulated and accelerated.29,30 In addition, these quantitative data will be helpful in managing and counseling families of afflicted patients.
References
Verheijen FW, Verbeek E, Aula N, et al. A new gene, encoding an anion transporter, is mutated in sialic acid storage diseases. Nat Genet. 1999;23:462–5.
Pertti A, Gahl WA. Disorders of free sialic acid storage. In: Valle D, editor. The online metabolic and molecular bases of inherited disease 2018. https://ommbid.mhmedical.com/content.aspx?sectionid=62655871&bookid=971&Resultclick=2. Accessed 15 March 2018.
Mancini GM, Beerens CE, Aula PP, et al. Sialic acid storage diseases. A multiple lysosomal transport defect for acidic monosaccharides. J Clin Invest. 1991;87:1329–35.
Aula P, Autio S, Raivio KO, et al. “Salla disease”: a new lysosomal storage disorder. Arch Neurol. 1979;36:88–94.
Tondeur M, Libert J, Vamos E, et al. Infantile form of sialic acid storage disorder: clinical, ultrastructural, and biochemical studies in two siblings. Eur J Pediatr. 1982;139:142–7.
Miyaji T, Echigo N, Hiasa M, et al. Identification of a vesicular aspartate transporter. Proc Natl Acad Sci USA. 2008;105:11720–4.
Prolo LM, Vogel H, Reimer RJ. The lysosomal sialic acid transporter sialin is required for normal CNS myelination. J Neurosci. 2009;29:15355–65.
Stroobants S, Van Acker NG, Verheijen FW, et al. Progressive leukoencephalopathy impairs neurobehavioral development in sialin-deficient mice. Exp Neurol. 2017;291:106–19.
Orphanet Report Series. Prevalence of rare diseases: bibliographic data. June 2017, no. 1. http://www.orpha.net/orphacom/cahiers/docs/GB/Prevalence_of_rare_diseases_by_alphabetical_list.pdf. Accessed 31 December 2017.
US Food and Drug Administration. Search orphan drug designations and approvals. 2017. https://www.accessdata.fda.gov/scripts/opdlisting/oopd/. Accessed 31 December 2017.
Mechler K, Mountford WK, Hoffmann GF, et al. Ultra-orphan diseases: a quantitative analysis of the natural history of molybdenum cofactor deficiency. Genet Med. 2015;17:965–70.
Zielonka M, Garbade SF, Kolker S, et al. Quantitative clinical characteristics of 53 patients with MPS VII: a cross-sectional analysis. Genet Med. 2017;19:983–8.
Zielonka M, Garbade SF, Kolker S, et al. A cross-sectional quantitative analysis of the natural history of Farber disease: an ultra-orphan condition with rheumatologic and neurological cardinal disease features. Genet Med. 2018;20:524–30.
von Elm E, Altman DG, Egger M, et al. The strengthening the reporting of observational studies in epidemiology (STROBE) statement: guidelines for reporting observational studies. Lancet. 2007;370:1453–7.
Hothorn T, Hornik K, Zeileis A. Unbiased recursive partitioning: a conditional inference framework. J Comput Graph Stat. 2006;15:651–74.
Ries M, Moore DF, Robinson CJ, et al. Quantitative dysmorphology assessment in Fabry disease. Genet Med. 2006;8:96–101.
Kahle D, Wickham H. ggmap: spatial visualization with ggplot2. R J. 2013;5:144–61.
Gimovsky AC, Luzi P, Berghella V. Lysosomal storage disease as an etiology of nonimmune hydrops. Am J Obstet Gynecol. 2015;212:281–90.
Helip-Wooley A, Kleta R, Gahl WA. Lysosomal free sialic acid storage disorders: Salla disease and ISSD. In: Barranger JAC-SM, editor. Lysosomal storage disorders. New York: Springer; 2007. p. 499–511.
Adams D, Gahl WA. Free sialic acid storage disorders. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews. Seattle, WA: University of Washington; 1993.
Hinderlich S, Weidemann W, Yardeni T, et al. UDP-GlcNAc 2-epimerase/ManNAc kinase (GNE): a master regulator of sialic acid synthesis. Top Curr Chem. 2015;366:97–137.
Mancini GM, Verheijen FW, Beerens CE, et al. Sialic acid storage disorders: observations on clinical and biochemical variation. Dev Neurosci. 1991;13:327–30.
Sidransky E. Gaucher disease: complexity in a “simple” disorder. Mol Genet Metab. 2004;83:6–15.
Accurso FJ, Rowe SM, Clancy JP, et al. Effect of VX-770 in persons with cystic fibrosis and the G551D-CFTR mutation. N Engl J Med. 2010;363:1991–2003.
Harutyunyan M, Huang Y, Mun KS, et al. Personalized medicine in CF: from modulator development to therapy for cystic fibrosis patients with rare CFTR mutations. Am J Physiol Lung Cell Mol Physiol. 2018;314:L529–L543.
Chiriboga CA, Swoboda KJ, Darras BT, et al. Results from a phase 1 study of nusinersen (ISIS-SMN(Rx)) in children with spinal muscular atrophy. Neurology. 2016;86:890–7.
Hua Y, Vickers TA, Okunola HL, et al. Antisense masking of an hnRNP A1/A2 intronic splicing silencer corrects SMN2 splicing in transgenic mice. Am J Hum Genet. 2008;82:834–48.
Hinderhofer K, Mechler K, Hoffmann GF, et al. Critical appraisal of genotype assessment in molybdenum cofactor deficiency. J Inherit Metab Dis. 2017;40:801–11.
Mechler K, Mountford WK, Hoffmann GF, et al. Pressure for drug development in lysosomal storage disorders—a quantitative analysis thirty years beyond the US Orphan Drug Act. Orphanet J Rare Dis. 2015;10:46.
Wagner JA, Dahlem AM, Hudson LD, et al. Application of a dynamic map for learning, communicating, navigating, and improving therapeutic development. Clin Transl Sci. 2018;11:166–174.
Acknowledgements
M.Z. received support from the Physician-Scientist Program at Ruprecht-Karls-University Heidelberg Faculty of Medicine.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
DISCLOSURE
The authors declare no conflicts of interest.
Rights and permissions
About this article

Cite this article
Zielonka, M., Garbade, S.F., Kölker, S. et al. A cross-sectional quantitative analysis of the natural history of free sialic acid storage disease—an ultra-orphan multisystemic lysosomal storage disorder. Genet Med 21, 347–352 (2019). https://doi.org/10.1038/s41436-018-0051-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-018-0051-3
Keywords
This article is cited by
-
Psychiatric symptoms in Salla disease
European Child & Adolescent Psychiatry (2023)
-
Lysosomes as a therapeutic target
Nature Reviews Drug Discovery (2019)
