
Abstract
Purpose
Sixty to seventy-five percent of individuals with rare and undiagnosed phenotypes remain undiagnosed after exome sequencing (ES). With standard ES reanalysis resolving 10–15% of the ES negatives, further approaches are necessary to maximize diagnoses in these individuals.
Methods
In 38 ES negative patients an individualized genomic–phenotypic approach was employed utilizing (1) phenotyping; (2) reanalyses of FASTQ files, with innovative bioinformatics; (3) targeted molecular testing; (4) genome sequencing (GS); and (5) conferring of clinical diagnoses when pathognomonic clinical findings occurred.
Results
Certain and highly likely diagnoses were made in 18/38 (47%) individuals, including identifying two new developmental disorders. The majority of diagnoses (>70%) were due to our bioinformatics, phenotyping, and targeted testing identifying variants that were undetected or not prioritized on prior ES. GS diagnosed 3/18 individuals with structural variants not amenable to ES. Additionally, tentative diagnoses were made in 3 (8%), and in 5 individuals (13%) candidate genes were identified. Overall, diagnoses/potential leads were identified in 26/38 (68%).
Conclusions
Our comprehensive approach to ES negatives maximizes the ES and clinical data for both diagnoses and candidate gene identification, without GS in the majority. This iterative approach is cost-effective and is pertinent to the current conundrum of ES negatives.
Similar content being viewed by others


Introduction
Exome sequencing (ES) has transformed the diagnostic approach to rare and undiagnosed Mendelian phenotypes, with diagnosis rates of 25–50%.1,2,3,4 However, 50–75% of individuals remain undiagnosed after ES (ES negatives). The next steps after a negative ES are currently limited. Some commercial laboratories offer one free ES reanalysis and this can provide a diagnosis in 10–15%, with the majority (~70%) occurring due to interim new gene–disease associations.5, 6 Other studies have reported diagnosis rates of 15–36% with ES reanalyses: although the raw data are reanalyzed, the diagnoses are mostly related to resequencing singletons as trios, looking for copy-number variants (CNV), literature reports, and case matching through platforms such as Matchmaker exchange.7, 8 Resequencing is reported to result in ~15% new molecular diagnoses, due to addition of family members and new gene–disease associations.9 We reported that reanalysis of ES can improve the diagnostic yield due to phenotypic characterization, improved exome coverage, realignment, and variant calling, in addition to new disease gene discovery,10 and careful phenotyping leading to targeted molecular testing can detects variants missed by ES.11 Genome sequencing (GS) can be an option for ES negative patients, with its ability to detect variants in noncoding regions, uniform coverage, and better detection of structural variants; ~15% of variants missed by ES may be detected by GS.12,13,14 However, GS is not widely available clinically and is not covered by third party payers and thus ES remains the standard diagnostic approach to rare Mendelian phenotypes.
There are many reasons why ES may miss variants of interest. Firstly, the underlying genetic etiology may be non-Mendelian and thus not amenable to ES (e.g., complex diseases). Secondly, the underlying disorder may be Mendelian, but due to variants undetectable with ES technology (e.g., trinucleotide repeats). Finally, variants that should be tractable to ES may still not be detected or reported due to variants not being detected or not recognized as disease-causing.10 This may occur due to (1) analytical factors/differences (e.g., difficult regions of exome, different quality filters);15, 16 (2) knowledge gaps since initial ES (e.g., evolving phenotypes, gene–disease relationships not well established);17, 18 (3) interpretation/reporting differences between labs (e.g., variant not reported due to poor phenotypic fit).11, 19
The Undiagnosed Diseases Network (UDN) (https://undiagnosed.hms.harvard.edu) is a nationwide National Institutes of Health (NIH)-funded research study that accepts patients with intractable phenotypes for further diagnostic resolution. Genomic sequencing is a major component of the UDN, because most undiagnosed and rare diseases (~85%) are believed to be genetic.20 At the Duke/Columbia clinical site of the UDN, we observed that the majority (~60%) who enter the study have a negative ES result through prior commercial or research sequencing. In comparison with previous studies, these individuals are among the most challenging, with the majority having undergone trio ES prior to the UDN and in some instances, an ES reanalysis as well. We thus devised a systematic approach to resolving these phenotypes, including reanalyses of the ES data with our innovative and agnostic approach in parallel to phenotyping and then utilizing the information from these iteratively. If the phenotype was specific enough to warrant targeted molecular tests, these were pursued and if still not resolved, GS was utilized. Our study provides an integrated genomic–phenomic approach to resolving ES negative individuals that extends well beyond just ES reanalyses.
Materials and methods
The study was performed under protocols approved by the Institutional Review Boards of Duke University Medical Center and the National Human Genome Research Institute (NHGRI).
Demographics
Thirty-eight individuals with a pre-UDN negative ES evaluated at the Duke/Columbia UDN clinical site from September 2015 to October 2017 were included. Nineteen patients (50%) were male, 29 (77%) were Caucasians, with 2 (5%) African Americans, 5 (13%) Asians, 2 (5%) Others, and 6 (16%) were Hispanic. The mean age was 7.07 ± 5.82 years, ranging from 0 to 26 years. The mean age of onset of illness was 0.51 ± 1.04 years and the mean duration of illness was 6.35 ± 5.59 years. The organ system most often involved was the nervous system (58%) with the musculoskeletal and gastrointestinal systems being the next most frequent at 7% each (Table S1). The time to diagnosis was 5.76 ± 5.22 months (0–23 months) in those who obtained a diagnosis (n = 21), compared with 10.83 ± 5.2 months, for declaration of no diagnosis in 12 individuals (t = 2.21, p < 0.05, Fig. S1).
Details of prior ES
Pre-UDN ES had been performed in 37 individuals and a pre-UDN GS in 1. A negative ES/GS was operationalized as an ES/GS report that was nondiagnostic and had either (1) no variants of interest, (2) variants of uncertain significance (VUS) in a known disease-causing gene, or (3) variants in candidate genes/genes of uncertain significance not associated with human disease. These pre-UDN variants are in Table S2.
Commercial ES had been performed in 22 individuals (59%), research ES in 13 (35%), and 2 (6%) had undergone both clinical and research ES (individual 23 had clinical GS). The majority were trios (33/38, 86%), 2 were quartets, 2 were duos (parent–child), and 1 was a singleton. The pre-UDN sequencing had occurred from 2012 to 2016, with the majority (36/38, 95%) occurring after 2012. One ES reanalysis prior to UDN entry had occurred in 18/38 (48%) individuals, 2.11 ± 1.07 years (1–4 years) after the initial ES.
Process for evaluation of ES negative individuals
We began with simultaneous ES data reanalyses and phenotyping. These data were iteratively used to derive variants of interest that could be pursued further for diagnoses. If the reanalyses found variants that were likely pathogenic, then the phenotyping was customized to capture clinical manifestations related to that particular disorder. If the phenotyping suggested specific conditions in the differential diagnosis, the ES data were reexamined for pertinent genes. Then, if no variants were detected and the clinical suspicion for a particular disorder was high, direct Sanger sequencing/deletion-duplication testing and/or biochemical testing was pursued. GS was utilized when these procedures did not result in resolution.
UDN phenotyping
Thirty-seven individuals underwent phenotyping (individual 23 died after acceptance and prior to evaluation). This included customized clinical consultations, imaging, procedures, and laboratory tests, during a 1-week visit to the Duke campus. Clinical consultations were the most often obtained (median = 3), with radiological, laboratory tests, and procedures being performed as needed (median = 1).
Review of other prior pertinent results
A chromosomal microarray that was at least at the level of an oligonucleotide array was available on 36/38 individuals and reviewed; no CNVs that could explain the individuals’ features were evident. Regions of homozygosity on the array, if present, were utilized to identify autosomal recessive genes of interest. Other pre-UDN laboratory test results were reviewed, but details are beyond the scope of this publication.
ES reanalyses
FASTQ files were obtained directly, or generated with data from the pertinent laboratory in 35/38 individuals. In three individuals (24, 27, and 23) raw ES/GS data could not be obtained. Primary alignment was performed with the DRAGEN platform.21 Duplicate removal was performed using Picard tools and index realignment and variant calling conducted with GATK v3.6. Variants were annotated using Clin-Eff with Ensembl-GRCh37.73. Our bioinformatics is agnostic in its approach, utilizing the innovative tools developed by our group. The Residual Variation Intolerance Score (RVIS) assesses whether genes have accumulated common functional variation; subRVIS applies the RVIS approach to subregions of genes and captures regional changes due to isoform inclusion/exclusion of exons, and/or by gene domain.22, 23 Novel genotypes were filtered into tier 1 and tier 2 variants. Tier 1 variants were strictly filtered for quality and control observations in public databases (ExAC, gnomAD,24 and EVS25), and 13,000 internal controls. Tier 1 variants were further prioritized: hot zone variants (polyphen 2 score >0.95 in an intolerant gene with an RVIS or a sub-RVIS score <25) were predicted damaging in an intolerant gene. We highlighted hot zone variants in known OMIM genes, or mouse essential genes. We also highlighted loss of function (LoF) variants that are in genes with known pathogenic LoF variants or reported as haploinsufficient by ClinGen,26 or LoF intolerant by high pLI score and estimated conservation/constraint of a variant site with the Genomic Evolutionary Rate Profiling (GERP) score.27 We curated ClinVar, HGMD, and internal cases to annotate all variants previously reported pathogenic. Tier 2 variants had less strict filters for quality and control observations, but required that a variant is a known or expected pathogenic variant. This allowed pathogenic variants that might otherwise be filtered due to noise in the control data sets. De novo, newly homozygous, newly hemizygous, and compound heterozygous variants were identified. All coding and intron/exon boundary (up to 8 bp) variants were also considered. An inheritance naïve filter was also applied to identify any variants that may be incompletely penetrant or mosaic in the parent. For genes known to be disease-associated, we also used the American College of Medical Genetics and Genomics (ACMG) criteria for variant classification (Table S1). In select cases, CNV analysis was performed with the target coverage and segmentation tools in GATK 4. These rely on normal samples sequenced on the same sequencing platform. With reanalysis these controls were not always available.
Exome sequencing
Two individuals (15 and 22, Table S1) had a repeat ES since the prior trio ES had been performed in early 2012, when ES capture kits were more incomplete. These were performed at the Baylor Miraca sequencing core of the UDN, using methodology and analyses previously published.28, 29
Genome sequencing
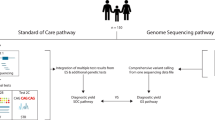
UDN GS was performed by the HudsonAlpha UDN sequencing core on 27 individuals (26 trios, one quartet), with methodology and analyses as previously published.30 The 27 individuals included 17 whose ES reanalyses through our study was negative and individual 23 who had a pre-UDN negative GS, as well as 9 other individuals whose GS was done in parallel with the ES reanalyses (Fig. 1 and Table S1).

Flowchart illustrating the approach to the exome sequencing (ES) negatives and the resolution with the different modalities
Communication with laboratories regarding ES negative results
When new variants were detected, we corresponded with the pertinent laboratory to discuss the reasons for the variant not being detected or not prioritized previously. This information is in the relevant tables (Tables 1–4 and Table S1).
Determination of diagnoses
The genomic and clinical information was combined for diagnostic interpretation by consensus. The UDN has created categories of diagnoses, recognizing that it is difficult to determine the certainty of diagnosis in rare phenotypes and that the certainty may change over time. Of the four categories of Certain, Highly Likely, Tentative, and Low, we used the first three to classify the diagnoses in the ES negatives in this study. Further considerations in this rubric are the method used to achieve the diagnosis (e.g., genomic sequencing, directed testing based on phenotype or clinical grounds), the mechanistic characterization of disease pathology, the degree to which the diagnosis explains the phenotypes of the patients, and consequences of the diagnoses. Whenever pertinent, variants were confirmed by Sanger sequencing/multiplex ligation-dependent probe amplification (MLPA)/exon array, prior to communication to the individuals and their families. When bioinformatically compelling variants in novel genes were identified, these were categorized as candidate genes. If further avenues such as GeneMatcher and functional studies led to the determination that they were new disease genes, they were then classified as a diagnosis (Certain, Highly Likely, or Tentative, depending on the strength of the supporting evidence).
Results
Overall, 18/38 (47%) individuals received Certain (n = 12) or Highly Likely diagnoses (n = 6) and 3 (8%) received Tentative diagnoses. Candidate genes were identified in 5 (13%) individuals. In total, we identified diagnoses/potential leads in 26/38 (68%) individuals (Table 1). In the individuals with a Certain or Likely Diagnoses (excluding the two with clinical diagnoses only), 8 had de novo autosomal dominant variants, 6 had biallelic autosomal recessive variants, 1 had an inherited autosomal dominant variant, and 1 had a de novo X-linked dominant variant (Tables 2, 3, and S1).
ES reanalyses
In 8/35 (23%) individuals, a Certain or Highly Likely diagnosis was made and a ninth individual received a Tentative diagnosis after ES reanalyses (Table 2, Fig. 1). The reasons for a pre-UDN negative ES in these individuals are listed in Tables 2, 4, and S1. Other variants detected in our ES reanalyses are in Tables S2 and S3. Overall, in these nine individuals, in one instance there was an interim literature report of a new gene–disease association (EFL1, Table 2). Except for the homozygous CNV in NPHP1 (individual 3, Table 2) which may have been easier to detect by GS, all the variants were of the type that are tractable by ES.
Integration of phenotype with genomic data from ES reanalyses
Genomic findings directed the phenotyping and the phenotypic information led to the examination of specific genes. For example, for individual 1 (Table 2 and S1), the EFL1 gene variant was detected just as phenotyping was beginning; further evaluations resulted in finding hematological, hepatic and pancreatic abnormalities consistent with the Shwachman–Diamond (SDS)-like syndrome associated with EFL1.31 Conversely, phenotyping by the epileptologist led to a recommendation to examine the CACNA1A gene in individual 2 (Table 2 and S1) and a likely pathogenic variant was detected on manual inspection of the gene and also on reanalyses through the pipeline. There were no significant differences in whether the pre-UDN ES was clinical or research based, among the Certain and Highly Likely diagnoses (χ2 = 0.46, p > 0.05).
Phenotype guided diagnoses
A strong clinical suspicion of specific disorders occurred in two individuals. Targeted molecular testing then led to pathogenic variants that had been missed on the pre-UDN ES, as published previously11 (Table 3, Fig. 1). A VUS in a known disease-causing gene in individual 17 was reinterpreted because she and her mother (who also has this variant) have pathognomonic features of the HEPACAM related disorder on further phenotyping. A CNV was established as being diagnostic for some features in individual 18, based on an interim literature report of this CNV being associated with features that overlapped his.32 In individuals 15 and 19 clinical diagnoses were conferred according to the UDN diagnostic rubric, due to their clinical features being so exactly consistent with a specific disorder, that the lack of molecular confirmation after all testing did not take away the diagnoses (Table 3 and S1).
Updated ES
Individual 14 (Table 3 and S1) had a repeat trio ES and a candidate gene variant in ASXL2 was proven to be associated with a new neurodevelopmental disorder.17 Individual 22 had a negative repeat ES and was subsequently diagnosed on GS due to a structural variant.
Genome sequencing (GS)
Three Certain/Highly Likely diagnoses were obtained in 3/18 (16%) individuals who underwentGS, after all other modalities to achieve a diagnosis had failed. All three diagnoses were due to structural variants that had not been detected on pre-UDN ES, due to the difficulty in detecting indels larger than 15 bp with ES.12, 33 Two others obtained a Tentative diagnosis and one candidate gene was identified on GS (Tables 1, 3, and S1). Interestingly, GS was also pursued in nine individuals (Fig. 1, Table S1) whose ES reanalyses were in progress. In all nine individuals, it was the ES reanalyses that led to either a diagnosis or a candidate gene, with the GS not prioritizing these variants (reasons in Tables 1–4).
New gene–disease associations
Two new gene–disease associations were established (ASXL2 and NACC1)17, 18 after initial identification as candidate genes. For two other genes identified as candidates we have evidence through further functional studies, animal modeling, and networking through GeneMatcher34 to judge these as disease-associated (AGTPBP1, IRF2BPL, publications in progress) (Table 2 and S1).
Candidate Genes
Our ES reanalyses identified four new candidate genes. GS did identify a fifth candidate gene, TBX2, and all are being studied currently (Fig. 1, Table 3 and Table S1).
Secondary findings
Two individuals were found on GS to have secondary findings. The father of individual 24 was homozygous for the common pathogenic variant in the HFE gene for hemochromatosis and individual 15 had a pathogenic variant in a long QT syndrome gene KCNE1 (Table S1). These were communicated to the families with management recommendations and genetic counseling.
Phenotypes of the ES negatives who remain undiagnosed
Twelve of the 38 individuals remain without a diagnosis or candidate genes. There were no significant demographic differences between these individuals and the others. Their manifestations were less often within the nervous system (41%) compared with 65% in the 26 individuals wherein a diagnosis or a potential lead was available; although this difference was not significant (Fisher’s exact test p > 0.05), we also observed that many of the 12 individuals had phenotypes that were representative of complex disorders (Table S1).
Discussion
A systematic approach to resolving diagnoses in ES negative individuals is a critical need, as the genomics community is increasingly utilizing ES in routine clinical practice and yet 50–75% of individuals remain without a diagnosis. We demonstrate that careful consideration of the phenotypic features, combined with innovative agnostic bioinformatics ES reanalysis, targeted molecular testing, and subsequent GS results in a significant number of the ES negatives being resolved (47%), with an additional ~20% obtaining tentative diagnoses or candidate genes. Our experience is that GS is highly effective in detecting structural variants, making it an important adjunct approach to ES negatives. However, mining ES data to maximize its potential and utilizing phenotype directed targeted testing can detect/prioritize variants not reported (due to analytical factors, knowledge gaps, and variability in laboratory reporting), so that the more expensive option of GS may be minimized (>80% of the molecular diagnoses we made were made without GS).
Prior studies on ES negatives have employed various approaches, including reanalyses of the raw ES data (sometimes with more relaxed filters), moving from singleton to trio sequencing to detect de novos and compound heterozygous variants, utilizing CNV analyses, considering the interim literature for new gene–disease associations and networking to identify additional patients; these procedures have yielded 10–36% additional diagnoses.5,6,7,8, 35 Our cohort was particularly challenging because the majority had been sequenced as trios (88%), with almost all having a negative pre-UDN CNV analysis (94%) and a substantial number (48%) entering the study with one negative ES reanalysis. Thus, many logical next steps were not avenues that we could pursue. Despite this, our systematic and comprehensive approach resulted in ~70% of the individuals obtaining diagnoses or potential leads that could be pursued further. Our ES reanalyses alone were highly effective in providing diagnostic resolution in approximately 25% of the ES negative individuals in this study. Only two diagnoses were facilitated by new disease gene reports in the interim literature and all diagnoses were achieved without the relatively easy step of moving from a singleton to a trio. Utilization of networking platforms such as GeneMatcher or Matchmaker Exchange34, 35 did facilitate candidate gene follow-up.17, 18
Our approach enabled us to identify variants that had been not been previously reported. Due to our innovative bioinformatics tools such as RVIS and our ranking of variants into tiers we were able to overcome analytical factors to select bioinformatically compelling variants. Capturing phenotypic changes allowed us to bridge knowledge gaps, resulting in identification of significant variants. Indeed analytical factors and knowledge gaps were the major reasons (70%) for a pre-UDN negative ES (Table 4). Variability in laboratory reporting resulted in nonreporting of significant variants when they did not fit the reported phenotype; this has implications for clinical practice as diagnoses can be missed and phenotypic expansion of a disorder may go unrecognized.
An important component of our systematic approach is to phenotype the ES negative individuals in parallel with the ES reanalyses. Phenotyping is also critical in solving ES negatives without automatically resorting to GS. When the clinical phenotypes are specific enough to be suggestive of one or a few disorders, targeted molecular testing, such as Sanger sequencing, can be effective in determining variants that can be difficult to detect on ES, and is also cost-effective.11 Finally, we were able to confer clinical diagnoses in two individuals, even in the absence of molecular conformation, because unmistakable pathognomonic phenotypic features of a specific disorder were present. Such clinical diagnoses, when prudently made with irrefutable findings, provide a guide to the families and enable reasonable genetic counseling and estimates of reproductive risk, even as efforts to find a molecular basis continue.
The role of GS in current diagnostics of rare and undiagnosed phenotypes is still being determined. In a cohort of individuals with intellectual disabilities and a negative ES, GS led to diagnosis in ~40% due to detection of de novo and structural variants in the exome;12 in such earlier studies, limitations of older ES capture kits may have led to coding variants not being detected. Other publications have estimated that 15–17% of additional diagnoses variants are made on GS, mostly due to detection of variants not amenable to ES.13, 14 Interestingly, in our cohort, GS led to a similar rate of Certain and Likely diagnoses (16%) in the ES negatives and all were due to structural variants that would be not be easily amenable to ES or chromosomal microarrays. We acknowledge that the majority of variants that we detected in this study would have been amenable to GS, but several patients (n = 9) who underwent GS were ultimately resolved by our systematic approach and not by GS. Varying reasons are operative for the negative GS in these individuals, such as the UDN GS laboratory not reporting variants that do not fit the described phenotype well and not reporting variants in genes of uncertain significance. This further emphasizes the value of using different pipelines in reinterpreting raw data on ES negatives. Establishing collaborations with researchers at their institutions or outside may enable clinicians to utilize a different bioinformatics pipeline for the reanalyses of ES data.
Twelve individuals in our study have no diagnosis or candidate genes, despite all efforts. A few of these individuals have phenotypes wherein the etiology could be complex (e.g., inflammatory bowel disease, autism, and recurrent fevers) and we propose that such disorders are difficult to solve by sequencing, unless larger cohorts with similar manifestations are accumulated and studied.
In conclusion, a systematic and comprehensive iterative approach to ES negatives that includes ES reanalysis, careful phenotyping, targeted testing, and in select cases GS can result in a high rate of resolution. We recommend that with the high cost, relatively low incremental yield over ES, and complexity of analyses, that GS be utilized only after ES data have been extensively mined and combined with the phenotypic data to maximize its yield. Many aspects of our approach can be implemented in practice. Commercial laboratories could adopt an agnostic approach (which could be easily automatable) to the raw data in ES negatives, so that variants that may be otherwise be filtered out due to stringent settings or phenotypic mismatch would be detected. Clinicians can also update laboratories about interim changes or atypical aspects of the phenotypes and ask about bioinformatically compelling variants that may have been initially unreported due to phenotypic mismatch. Utilizing targeted testing such as Sanger sequencing for disorders that are high in the differential diagnosis is useful; these variants may have been missed on ES, due to various analytical factors, as illustrated by Individuals 16 and 20 in this study. Additionally, considering disorders that are not amenable to ES (e.g., epigenetic disorders) and obtaining a chromosomal microarray (if not previously done) are useful approaches to ES negatives. Finding additional cases through networking such as GeneMatcher are also feasible in clinical practice. As the genomics community faces the challenge of the ES negatives, approaches such as ours provide viable avenues to maximize their resolution.
References
Lee H, Deignan JL, Dorrani N, et al. Clinical exome sequencing for genetic identification of rare Mendelian disorders. JAMA. 2014;312:1880–7.
Gilissen C, Hoischen A, Brunner HG, Veltman JA. Unlocking Mendelian disease using exome sequencing. Genome Biol. 2011;12:228.
Need AC, Shashi V, Hitomi Y, et al. Clinical application of exome sequencing in undiagnosed genetic conditions. J Med Genet. 2012;49:353–61.
Yang Y, Muzny DM, Xia F, et al. Molecular findings among patients referred for clinical whole-exome sequencing. JAMA. 2014;312:1870–9.
Wenger AM, Guturu H, Bernstein JA, Bejerano G. Systematic reanalysis of clinical exome data yields additional diagnoses: implications for providers. Genet Med. 2017;19:209–14.
Smith ED, Radtke K, Rossi M, et al. Classification of genes: standardized clinical validity assessment of gene-disease associations aids diagnostic exome analysis and reclassifications. Hum Mutat. 2017;38:600–8.
Eldomery MK, Coban-Akdemir Z, Harel T, et al. Lessons learned from additional research analyses of unsolved clinical exome cases. Genome Med. 2017;9:26.
Nambot S, Thevenon J, Kuentz P, et al. Clinical whole-exome sequencing for the diagnosis of rare disorders with congenital anomalies and/or intellectual disability: substantial interest of prospective annual reanalysis. Genet Med. 2018;20:645–54.
Gibson KM, Nesbitt A, Cao K, et al. Novel findings with reassessment of exome data: implications for validation testing and interpretation of genomic data. Genet Med. 2018;20:329–36.
Need AC, Shashi V, Schoch K, Petrovski S, Goldstein DB. The importance of dynamic re-analysis in diagnostic whole exome sequencing. J Med Genet. 2017;54:155–6.
Pena LDM, Jiang YH, Schoch K, et al. Looking beyond the exome: a phenotype-first approach to molecular diagnostic resolution in rare and undiagnosed diseases. Genet Med. 2018;20:464–9.
Gilissen C, Hehir-Kwa JY, Thung DT, et al. Genome sequencing identifies major causes of severe intellectual disability. Nature. 2014;511:344–7.
Taylor JC, Martin HC, Lise S, et al. Factors influencing success of clinical genome sequencing across a broad spectrum of disorders. Nat Genet. 2015;47:717–26.
Lionel AC, Costain G, Monfared N, et al. Improved diagnostic yield compared with targeted gene sequencing panels suggests a role for whole-genome sequencing as a first-tier genetic test. Genet Med. 2018;20:435–43.
Sims D, Sudbery I, Ilott NE, Heger A, Ponting CP. Sequencing depth and coverage: key considerations in genomic analyses. Nat Rev Genet. 2014;15:121–32.
Fang H, Wu Y, Narzisi G, et al. Reducing INDEL calling errors in whole genome and exome sequencing data. Genome Med. 2014;6:89.
Shashi V, Pena LD, Kim K, et al. De novo truncating variants in ASXL2 are associated with a unique and recognizable clinical phenotype. Am J Hum Genet. 2016;99:991–9.
Schoch K, Meng L, Szelinger S, et al. A recurrent de novo variant in NACC1 causes a syndrome characterized by infantile epilepsy, cataracts, and profound developmental delay. Am J Hum Genet. 2017;100:343–51.
Pabinger S, Dander A, Fischer M, et al. A survey of tools for variant analysis of next-generation genome sequencing data. Brief Bioinform. 2014;15:256–78.
Rare Disease Statistics. 2015.https://globalgenes.org. Global Genes, CA. Accessed January 30, 2018.
genome E. http://edicogenome.com/dragen-bioit-platform/. Illumina, CA. Accessed January 30, 2018.
Petrovski S, Wang Q, Heinzen EL, Allen AS, Goldstein DB. Genic intolerance to functional variation and the interpretation of personal genomes. PLoS Genet. 2013;9:e1003709.
Gussow AB, Petrovski S, Wang Q, Allen AS, Goldstein DB. The intolerance to functional genetic variation of protein domains predicts the localization of pathogenic mutations within genes. Genome Biol. 2016;17:9.
Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–91.
(ESP) NGESP. Exome Variant Server.: http://evs.gs.washington.edu/EVS/. University of Washington, WA. Accessed January 30, 2018.
Resource CG. https://www.clinicalgenome.org/. ClinGen. Accessed January 30, 2018.
Cooper GM, Stone EA, Asimenos G, Green ED, Batzoglou S, Sidow A. Distribution and intensity of constraint in mammalian genomic sequence. Genome Res. 2005;15:901–13.
Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24.
Yang Y, Muzny DM, Reid JG, et al. Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N Engl J Med. 2013;369:1502–11.
Bick D, Fraser PC, Gutzeit MF, et al. Successful application of whole genome sequencing in a medical genetics clinic. J Pediatr Genet. 2017;6:61–76.
Stepensky P, Chacon-Flores M, Kim KH, et al. Mutations in EFL1, an SBDS partner, are associated with infantile pancytopenia, exocrine pancreatic insufficiency and skeletal anomalies in a Shwachman-Diamond like syndrome. J Med Genet. 2017;54:558–66.
Vlaskamp DR, Rump P, Callenbach PM, et al. Haploinsufficiency of the STX1B gene is associated with myoclonic astatic epilepsy. Eur J Paediatr Neurol. 2016;20:489–92.
Caspar SM, Dubacher N, Kopps AM, Meienberg J, Henggeler C, Matyas G. Clinical sequencing: from raw data to diagnosis with lifetime value. Clin Genet. 2017;93:508–19.
Sobreira N, Schiettecatte F, Valle D, Hamosh A. GeneMatcher: a matching tool for connecting investigators with an interest in the same gene. Hum Mutat. 2015;36:928–30.
Philippakis AA, Azzariti DR, Beltran S, et al. The Matchmaker Exchange: a platform for rare disease gene discovery. Hum Mutat. 2015;36:915–21.
Acknowledgements
This work has received support by the National Institutes of Health (NIH) Common Fund through the Office of Strategic Coordination/Office of the NIH Director (U01HG007672, to Shashi V and Goldstein DB). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Undiagnosed Diseases Network
David R. Adams, Mercedes E. Alejandro, Patrick Allard, Euan A. Ashley, Mahshid S. Azamian, Carlos A. Bacino, Ashok Balasubramanyam, Hayk Barseghyan, Gabriel F. Batzli, Alan H. Beggs, Babak Behnam, Hugo J. Bellen, Jonathan A. Bernstein, Anna Bican, David P. Bick, Camille L. Birch, Devon Bonner, Braden E. Boone, Bret L. Bostwick, Lauren C. Briere, Donna M. Brown, Matthew Brush, Elizabeth A. Burke, Lindsay C. Burrage, Manish J. Butte, Shan Chen, Gary D. Clark, Terra R. Coakley, Joy D. Cogan, Cynthia M. Cooper, Heidi Cope, William J. Craigen, Precilla D’Souza, Mariska Davids, Jean M. Davidson, Jyoti G. Dayal, Esteban C. Dell’Angelica, Shweta U. Dhar, Katrina M. Dipple, Laurel A. Donnell-Fink, Naghmeh Dorrani, Daniel C. Dorset, Emilie D. Douine, David D. Draper, Annika M. Dries, David J. Eckstein. Lisa T. Emrick, Christine M. Eng, Gregory M. Enns, Ascia Eskin, Cecilia Esteves, Tyra Estwick, Liliana Fernandez, Carlos Ferreira, Paul G. Fisher, Brent L. Fogel, Noah D. Friedman, William A. Gahl, Emily Glanton, Rena A. Godfrey, David B. Goldstein, Sarah E. Gould, Jean-Philippe F. Gourdine, Catherine A. Groden, Andrea L. Gropman, Melissa Haendel, Rizwan Hamid, Neil A. Hanchard, Lori H. Handley, Matthew R. Herzog, Ingrid A. Holm, Jason Hom, Ellen M. Howerton, Yong Huang, Howard J. Jacob, Mahim Jain, Yong-hui Jiang, Jean M. Johnston, Angela L. Jones, David M. Koeller, Isaac S. Kohane, Jennefer N. Kohler, Donna M. Krasnewich, Elizabeth L. Krieg, Joel B. Krier, Jennifer E. Kyle, Seema R. Lalani, C. Christopher Lau, Jozef Lazar, Kimberly LeBlanc, Brendan H. Lee, Hane Lee, Shawn E. Levy, Richard A. Lewis, Sharyn A. Lincoln, Sandra K. Loo, Joseph Loscalzo, Richard L. Maas, Ellen F. Macnamara, Calum A. MacRae, Valerie V. Maduro, Marta M. Majcherska, May Christine V. Malicdan, Laura A. Mamounas, Teri A. Manolio, Thomas C. Markello, Ronit Marom, Martin G. Martin, Julian A. Martínez-Agosto, Shruti Marwaha, Thomas May, Allyn McConkie-Rosell, Colleen E. McCormack, Alexa T. McCray, Jason D. Merker, Thomas O. Metz, Matthew Might, Paolo M. Moretti, Marie Morimoto, John J. Mulvihill, Jennifer L. Murphy, Donna M. Muzny, Michele E. Nehrebecky, Stan F. Nelson, J. Scott Newberry, John H. Newman, Sarah K. Nicholas. Donna Novacic, Jordan S. Orange, J. Carl Pallais, Christina GS. Palmer, Jeanette C. Papp, Neil H. Parker, Loren DM. Pena, John A. Phillips III, Jennifer E. Posey, John H. Postlethwait, Lorraine Potocki, Barbara N. Pusey, Chloe M. Reuter, Amy K. Robertson, Lance H. Rodan, Jill A. Rosenfeld, Jacinda B. Sampson, Susan L. Samson, Kelly Schoch, Molly C. Schroeder, Daryl A. Scott, Prashant Sharma, Vandana Shashi, Edwin K. Silverman, Janet S. Sinsheimer, Kevin S. Smith, Rebecca C. Spillmann, Joan M. Stoler, Nicholas Stong, Jennifer A. Sullivan, David A. Sweetser, Queenie K.-G. Tan, Cynthia J. Tifft, Camilo Toro, Alyssa A. Tran, Tiina K. Urv, Zaheer M. Valivullah, Eric Vilain, Tiphanie P. Vogel, Daryl M. Waggott, Colleen E. Wahl, Nicole M. Walley, Chris A. Walsh, Jijun Wan, Michael F. Wangler, Patricia A. Ward, Katrina M. Waters, Bobbie-Jo M. Webb-Robertson, Monte Westerfield, Matthew T. Wheeler, Anastasia L. Wise, Lynne A. Wolfe, Elizabeth A. Worthey, Shinya Yamamoto, Yaping Yang, Amanda J. Yoon, Guoyun Yu, Diane B. Zastrow, Chunli Zhao, Allison Zheng
Author information
Authors and Affiliations
Consortia
Corresponding author
Ethics declarations
Disclosure
David Goldstein is a founder of and holds equity in Pairnomix and Praxis, serves as a consultant to AstraZeneca, and has research supported by Janssen, Gilead, Biogen, AstraZeneca, and UCB. The remaining authors declare no conflicts of interest.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Shashi, V., Schoch, K., Spillmann, R. et al. A comprehensive iterative approach is highly effective in diagnosing individuals who are exome negative. Genet Med 21, 161–172 (2019). https://doi.org/10.1038/s41436-018-0044-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-018-0044-2
Keywords
This article is cited by
-
Reanalysis of whole-exome sequencing (WES) data of children with neurodevelopmental disorders in a standard patient care context
European Journal of Pediatrics (2023)
-
The diagnostic yield of exome sequencing in liver diseases from a curated gene panel
Scientific Reports (2023)
-
Participation in a national diagnostic research study: assessing the patient experience
Orphanet Journal of Rare Diseases (2023)
-
Overwhelming genetic heterogeneity and exhausting molecular diagnostic process in chronic and progressive ataxias: facing it up with an algorithm, a gene, a panel at a time
Journal of Human Genetics (2020)
-
A three-year follow-up study evaluating clinical utility of exome sequencing and diagnostic potential of reanalysis
npj Genomic Medicine (2020)
