
Abstract
Purpose
To improve methods for predicting the impact of missense variants of uncertain significance (VUS) in BRCA1 and BRCA2 on protein function.
Methods
Functional data for 248 BRCA1 and 207 BRCA2 variants from assays with established high sensitivity and specificity for damaging variants were used to recalibrate 40 in silico algorithms predicting the impact of variants on protein activity. Additional random forest (RF) and naïve voting method (NVM) metapredictors for both BRCA1 and BRCA2 were developed to increase predictive accuracy.
Results
Optimized thresholds for in silico prediction models significantly improved the accuracy of predicted functional effects for BRCA1 and BRCA2 variants. In addition, new BRCA1-RF and BRCA2-RF metapredictors showed area under the curve (AUC) values of 0.92 (95% confidence interval [CI]: 0.88–0.96) and 0.90 (95% CI: 0.84–0.95), respectively. Similarly, the BRCA1-NVM and BRCA2-NVM models had AUCs of 0.93 and 0.90. The RF and NVM models were used to predict the pathogenicity of all possible missense variants in BRCA1 and BRCA2.
Conclusion
The recalibrated algorithms and new metapredictors significantly improved upon current models for predicting the impact of variants in cancer risk–associated domains of BRCA1 and BRCA2. Prediction of the functional impact of all possible variants in BRCA1 and BRCA2 provides important information about the clinical relevance of variants in these genes.
Similar content being viewed by others

Introduction
Pathogenic variants in BRCA1 and BRCA2 account for 20–25% of hereditary breast and ovarian cancer,1 5–10% of breast cancers,2 and up to 15% of ovarian cancers.3 While most known pathogenic variants in these genes truncate the encoded proteins, missense variants can also predispose to cancer. More than 90% of missense variants in public databases4 identified by clinical genetic testing are listed as variants of uncertain significance (VUS).5 Missense variants with definitive pathogenic or neutral status can inform clinical management, prevention, and treatment. Thus, accurate methods to establish variant pathogenicity are needed.
Family-based studies yielding likelihoods of pathogenicity, based on segregation of variants with cancer and personal and family history of cancer, are established methods for determining pathogenicity of variants in BRCA1 and BRCA2. However, few missense variants have been clinically annotated by this method owing to the limited availability of family-based data. Similarly, functional assays6,7 with established specificity and sensitivity for known pathogenic and neutral BRCA1 or BRCA2 variants have been used alone or in combination with family-based segregation data to infer pathogenicity.8 However, classification of all possible variants by functional assays is unlikely. Alternatively, the clinical relevance of variants can be assessed using sequence-based in silico prediction models, which can be applied to all possible missense VUS in these genes. Given the large number of unique VUS identified in BRCA1 and BRCA2, in silico prediction models will need to be incorporated in models that aim to predict the pathogenicity of VUS in these genes. Most commonly used prediction tools such as SIFT,9 PolyPhen,10 GERP,11 Align-GVGD,12 and CADD13 have been developed using large-scale databases such as the Human Gene Mutation Database (HGMD)14 or ClinVar.4 While functional assays can outperform these computational predictions of damage,6,15 development and/or calibration of in silico prediction models using well-characterized functional data from validated assays is expected to improve variant annotation.
In this study, homology-directed repair (HDR)6 functional data from 207 BRCA2 variants and transcriptional integrity16,17 data for 248 BRCA1 variants were used to evaluate the performance of existing in silico algorithms. Sensitivity and specificity of the algorithms were optimized by defining more accurate thresholds, and by newer high-performance random forest (RF) and naïve voting method (NVM) predictors. We show that optimization for one gene leads to poor performance when applied to the other, highlighting the importance of different gene-specific features for prediction accuracy.
Materials and methods
BRCA1 transcription integrity assay
Results from functional studies of variants in the BRCT domains of BRCA1 using a transcription integrity assay have been reported previously.16,17 The sensitivity and specificity of this assay for missense variants in the BRCT domains of BRCA1 have been estimated at 100% (sensitivity, 95% confidence interval [CI]: 75−100%; specificity, 95% CI: 83−100%).16 The 95% probability of pathogenicity and neutrality from the VarCall two-component mixture model for classification of BRCA1 missense variants8 was used to define 61 pathogenic, 21 indeterminate (partial effect on function), and 166 neutral variants (total of 248). These data were used to define BRCA1 activity.
BRCA2 HDR assay
A cell-based homology directed DNA repair activity assay was used to assess the influence of missense variants in the DNA binding domain of BRCA2 on protein activity.6 In brief, BRCA2 activity in brca2 deficient V-C8 cells expressing mutant forms of full-length BRCA2 was measured with a DR-GFP (direct repeat-green fluorescent protein) reporter plasmid after induction of a DNA double-strand break using the I-Sce1 enzyme. The V-C8 hamster lung fibroblast cell line was a gift from Dr. Margaret Zdzienicka. Cells were verified by genotyping in the Mayo Clinic Medical Research Facility and routinely tested for mycoplasma contamination. The sensitivity and specificity of this assay for damaging missense variants in the DNA binding domain (DBD) of BRCA2 has previously been estimated at 100% (sensitivity, 95% CI: 79−100%; specificity, 95% CI: 93−100%) using 21 known neutral and 13 known pathogenic variants6,18,19,20. Results from 68 variants were combined with previous results from 139 previously characterized variants for a total of 207.
Damaging missense prediction tools
dbNSFP version 3.0a21 was downloaded and converted into a BioR catalogue22 to annotate variants. Align-GVGD12 was accessed online. CAROL and CONDEL scores were gathered from Variant Effect Predictor (VEP).23
Optimized thresholds
Analyses included damaging, indeterminate, and neutral variants. Indeterminate variants were included in the neutral category (scenario 1). An alternative approach, in which indeterminate variants are included in the pathogenic category (scenario 2) is provided in Supplemental Materials. Optimal thresholds for individual predictive algorithms that maximized sensitivity and specificity for damaging variants were derived using results from the BRCA1 transcriptional integrity assay and BRCA2 HDR assay, individually (Fig. S1 and S2). Matthews correlation coefficients (MCCs) were calculated for each resulting binary classification relative to the functional assay standards.24 The areas under the curve (AUCs) were estimated and reported with 95% confidence intervals using the DeLong error method. Receiver operating characteristic (ROC) analyses were performed using the package optimalCutpoints25 for R software (v3.3.3; http://www.R-project.org).
Naïve voting method (NVM) models
For each gene, a training set (a random sample of ~50% of the variants for each gene) and a test set (the remaining ~50%) were constructed using the sample function in R. The training set was used to determine the optimal number of individual prediction algorithms in the NVM model based on the maximal MCC. Starting with the individual prediction algorithm with the highest MCC, the prediction algorithm with the highest individual MCC among the models not previously chosen was added iteratively until the optimal numbers of prediction algorithms were included. If both a raw score (Score) and rank score (RankScore) for an algorithm were available, then only the RankScore was utilized. The NVM models and thresholds developed in the training sets were validated in the test sets. The MCC and other performance statistics were also recalculated across the entire data sets (training and test combined) to be consistent with reporting of other models. Lollipop plots were generated with lollipops (v1.2, https://doi.org/10.5281/zenodo.46184).
Random forest models
Random forest (RF) modeling utilized scores from each of the optimized individual prediction algorithms to identify the subset of prediction algorithms that maximized the accuracy of predicting damaging and nondamaging (indeterminate and neutral) variants in BRCA1 and BRCA2. The randomForest R package26 was used with settings of n = 500 trees and the number of predictor variables sampled as candidates at each split set to the recommended default of sqrt(p), where p is the number of predictor variables included in the model. For individual prediction algorithms available as both a Score and RankScore, only the RankScore was included in the random forest models. Variable importance was assessed using the mean decrease in accuracy resulting from exclusion of a given prediction model from the RF classifiers. Out-of-sample predictions on the probability scale were again derived for each model and used to estimate AUC, sensitivity, specificity, and MCC at optimized cut points for prediction of functional status.
Comparison to ClinVar
BRCA1 and BRCA2 classifications from ClinVar that were reviewed by an expert panel and had no conflicting interpretations were used. Pathogenic and likely pathogenic variants in ClinVar were grouped into the pathogenic (damaging) category, and variants annotated as benign or likely benign in ClinVar with no conflicting interpretations were defined as neutral (neutral).
Code availability
All code and data required to replicate all analyses are available on GitHub (https://github.com/Steven-N-Hart/NVM).
Results
Functional characterization of 68 novel BRCA2 missense variants
In this study, 68 BRCA2 variants from the BRCA2 DBD were evaluated using the HDR assay. Of these, 17 showed HDR fold change <1.66, with probabilities of pathogenicity >0.99 (Table 1, Fig. 1, Table S1), and 48 variants showed HDR >2.41 and probabilities of neutrality >0.99. Another three variants (p.I2672T, p.D2733V, and p.P3150L) displayed partial activity (HDR fold change >1.66 and <2.41) and were annotated as indeterminate variants (Fig. 1, Table S1). When combined with previously classified variants,6,18 69 were predicted deleterious (damaging), 21 were intermediate/partial (indeterminate), and 117 were predicted benign/neutral (neutral) (Table 1, Table S1).

The model-based HDR fold change with standard error (SE) is displayed on a logarithmic scale. The SE is included as a measure of the reproducibility of the HDR assay for each variant. Solid lines represent 99% probability of pathogenicity and 99% probability of neutrality (fold increase in GFP (+) cells <1.66 for damaging and fold increase in GFP (+) cells >2.41 for neutral). Dotted lines separate variants classified as deleterious, indeterminate, and neutral
Computational predictions
Sensitivity and specificity of 40 computational prediction models with previously established cut points for damaging variants were determined using the functional assay data for BRCA1 and BRCA2 missense variants (Tables S2, Table S3). These default thresholds yielded either high sensitivity with low specificity (e.g. BRCA2 SIFT score: sensitivity 100%, specificity <20%) or low sensitivity with high specificity (e.g., BRCA1 PROVEAN score: sensitivity <0.02%, specificity 100%), depending on the gene (Table S3).
To optimize the predictive ability of each model, thresholds that maximized sensitivity and specificity for damaging variants were defined separately for BRCA1 and BRCA2. For the purposes of predicting damaging, clinically relevant variants, models were generated by combining indeterminate with neutral variants (scenario 1). Performance characteristics and AUC values for optimized individual prediction models for BRCA1 and BRCA2 are shown in Table 2 and Figure S2. The best-performing individual models for BRCA1 incorporated conservation measures including deep interspecies protein alignments and physicochemical changes in amino acids (MetaSVM Score and RankScore,21 PERCH and PERCH_noMAF,27 Align-GVGD,12 Polyphen2Hvar Score and RankScore,10 and VEST3 Score and RankScore28). These models yielded AUCs >0.87, sensitivity and specificity >80%, and MCCs up to 0.68 (VEST3Score) (Table 2, Table S4). These results represented a major improvement in performance over results based on default thresholds (mean = 0.29) (Table S3). The best-performing models for BRCA2 were PERCH and PERCH_noMAF, MetaLR RankScore and Score, MetaSVM RankScore and Score, and VEST3 RankScore and Score. These yielded AUCs of 0.83–0.89, sensitivity and specificity >78% (85% for PERCH), and MCCs >0.53 (Table 2, Table S4), which were substantially improved over models using default parameters (MCC <0.42) (Table S3).
To assess whether metapredictor models improved prediction of the damaging variants for each gene, two new models were developed for both BRCA1 and BRCA2: (1) random forest (RF) classifiers of prediction methods were derived from the continuous outputs from the functional data (BRCA1-RF and BRCA2-RF), and (2) naïve voting methods (NVMs) were applied to optimized thresholds for each prediction model (BRCA1-NVM and BRCA2-NVM). CAROL29 and CONDEL30 predictors were not included in development of new BRCA2 models because prediction scores for 29 of 207 (14.0%) variants were not available. Only 12 of 248 (4.8%) BRCA1 and 1 of 207 (0.5%) BRCA2 variants were excluded from new model development due to missing data or conflicts between protein and DNA sequences (Table S2).
RF models
Random forest (RF) classifiers were used to evaluate the impact of excluding individual prediction methods on the accuracy of composite prediction models. VEST3 RankScore and Align-GVGD had the greatest impact on the accuracy of BRCA1-RF, whereas Mutation Assessor RankScore and PERCH had the greatest impact on BRCA2-RF. The BRCA1-RF model (threshold ≥0.298) (Table S4) showed the second highest AUC value of all models for BRCA1 (0.92, 95% CI: 0.88–0.96), with 86% sensitivity and 85% specificity. The BRCA1-RF model predicted 8 of 59 (13.6%) functionally impaired BRCA1 variants as neutral (false negatives), 12 of 21 (57.1%) functionally indeterminate variants as damaging, and 14 of 156 (9.0%) functionally intact neutral variants as damaging (false positives) (Table S2). Similarly, the BRCA2-RF model (threshold ≥0.371) (Table S4) had the highest AUC for BRCA2 (0.90, 95% CI: 0.84–0.95) (Table 2, Table S4) with 83% sensitivity and 82% specificity (Table S4, Fig. 2).

Higher values indicate increased classifier performance
NVM models
NVM models based on the optimal number of individual prediction algorithms for BRCA1 and BRCA2 variants were also developed. The optimal NVM for BRCA1, following training and validation (BRCA1-NVM combined) contained 13 prediction models (Table S5). BRCA1 variants were predicted damaging when ≥9 of the 13 models exceed their individual thresholds for damaging variants (Table S5). BRCA1-NVM yielded an AUC of 0.94 with sensitivity of 83% and specificity of 91%. The highest proportion of BRCA1 misclassifications involved variants with indeterminate function, with 9 of 21 (42.9%) annotated as damaging. In contrast, the optimal BRCA2-NVM (BRCA2-NVM combined) model after training and validation incorporated six prediction models with a threshold of ≥4 models predicting damaging variants (Table S5). This model yielded sensitivity of 82% and specificity of 87% (Table 2, Table S4, Table S5), with 14 functionally damaging variants predicted as neutral, and 18 indeterminate/neutral variants predicted as damaging. As with BRCA1, the false positive results were disproportionately enriched for indeterminate function with 6 of 21 (28.6%) misclassified. Overall, the predictive abilities of the RF and NVM models showed substantial improvement over individual in silico prediction methods using default parameters, and modest improvements over the best-performing individual in silico methods optimized at thresholds specific to BRCA1 and BRCA2.
Application of selected models to all possible missense variants in BRCA1 and BRCA2
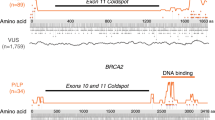
The RF and NVM models were used to assess the damaging potential of all theoretically possible missense substitutions resulting from single-nucleotide changes in BRCA1 and BRCA2, contingent on availability of prediction scores from all the individual methods contributing to each model (Table S2). Because a subset of the contributing prediction algorithms are in part based on nucleotide substitution rates, several missense variants caused by different nucleotide changes may have more than one predicted RF or NVM score. Using BRCA1-NVM, 7.1% of BRCA1 variants were predicted as damaging. Similarly, 2.6% of BRCA2 variants were predicted as damaging using BRCA2-NVM. However, marked enrichment for NVM predicted damaging variants was observed in known functional domains (Fig. 3). Analysis of the BRCA1 RING domain, predicted that 30–40% of all missense changes disrupt protein function. Similarly, 46% of all possible missense variants in the C-terminal BRCT domains and >20% in the larger C-terminal region (residue 1,660–1,810) were predicted damaging (Table S2). Interestingly, ~10% of all possible variants between amino acids 300 to 550, which have been associated with TP53,31 RAD50,32 and c-MYC31 interactions, were predicted damaging (Table S2). For BRCA2, only the region from residues 2,574 to 2,771 that contains the helical and OB1 domains of the DNA binding domain was predicted to have >20% damaging variants, although 10% of variants in OB3 were also predicted damaging (Fig. 3, Table S2). Few damaging missense variants were predicted in the OB2 domain. Similar results were obtained using the RF model (Table S2). Damaging pathogenic variations were not predicted in the N-terminus of BRCA2, containing the PALB2 interaction domain,33 possibly because of the small size of the interaction site.

The AAPOS x-axis represents the amino acid position, and the y-axis is the probability of a missense pathogenic variations being damaging from the naïve voting method (NVM) model. The lines were smoothed using a 50–amino acid sliding window
Discussion
Specific measures of BRCA1 and BRCA2 functional activity have been established as reliable measures of the functional impact and the likelihood of pathogenicity of variants in certain domains of BRCA1 and BRCA2.6,16 However, in the absence of functional studies of individual variants, in silico models that incorporate functional or structural data are often considered useful predictors of function. Here, existing models for prediction of damaging missense variants were recalibrated based on BRCA1 and BRCA2 functional data and were combined in metapredictor classifiers (NVM and RF). These metapredictors leveraged the strengths and weaknesses and improved upon many of the individual models for predicting the functional implications of missense variants in the cancer risk–associated domains of BRCA1 and BRCA2. We subsequently used these highly sensitive and specific models to annotate all missense variants from the BRCA1 and BRCA2 genes as damaging or neutral. Importantly, because the BRCA1 transcriptional integrity assay and the BRCA2 HDR assay used for calibration of the various prediction models have 100% sensitivity and specificity for clinically pathogenic variants in the BRCA1 BRCT and BRCA2 DNA binding domain domains, respectively, the models may also predict the clinical pathogenicity of missense variants in these domains. Whether prediction of functional effects in other parts of these proteins also reflects pathogenicity remains to be determined using additional pathogenic and neutral standards. Overall, these prediction models are likely to alter the interpretation of many VUS in BRCA1 and BRCA2, leading to improved clinical genetic testing, and perhaps improved risk management of patients found to carry VUS.
The current American College of Medical Genetics and Genomics guidelines for variant classification recommends that in silico evidence can be counted as supporting evidence for pathogenicity (or lack thereof) if all of the in silico programs tested agree on the prediction, whereas in silico evidence should not be used for classification if in silico predictions disagree. However, the guidelines do not recommend specific in silico methods, or indicate the number of methods that should be evaluated.34 This differs from the NVM model in two key areas. First, default thresholds of predictive models are not appropriate for BRCA1 and BRCA2 because the specificity is very low. The new thresholds for predictive models derived here should provide more accurate predictions of functional impact and therefore pathogenicity. Second, while using an ensemble of models is a rational strategy, requiring all models to be in agreement becomes overly stringent resulting in decreased performance (Figure S3 and Figure S4). Rather, the number of in silico models, the choice of which specific models, and the thresholds for those models that are required for an accurate consensus with both high sensitivity and specificity can vary by gene.
Effect of grouping indeterminate variants as either damaging or neutral
Generally, the performance of individual in silico prediction models, as well as the RF and NVM, were similar when indeterminate variants were grouped with either damaging or neutral. However, the performance of some of the known prediction methods was highly sensitive to indeterminate variant classification. Interestingly, the prediction methods that had the greatest difference in thresholds, depending on the incorporation of the indeterminate variants in the damaging or neutral categories, also had higher AUCs (e.g., PERCH, NVM, RF, MetaSVM) compared with those with no change in threshold (e.g., PolyPhen2HDiv, PolyPhen2HVar, MutationTaster Score), suggesting that the former methods are better predictors of indeterminate impact on function. However, the clinical relevance of the indeterminate variants in BRCA1 and BRCA2 is not well understood. Further understanding of function, pathogenicity, cancer risk, and associated refinement of thresholds for damage and pathogenicity for each functional assay may allow recalibration of the prediction models and improved prediction of clinically relevant BRCA1 and BRCA2 variants in the future.
Extending missense prediction outside established functional domains
When cross-referencing the NVM predictions with well-annotated ClinVar classifications, the predictions clustered in well-known domains, with damaging missense variants mostly restricted to BRCT and RING domains of BRCA1 and the DNA binding domain of BRCA212 (Fig. S5). The BRCA1-NVM prediction model clearly delineated both regions, with as many as 40% of missense variants in the RING domain and 50% in parts of the BRCT regions annotated as damaging. Interestingly, enrichment between amino acids 400–500 was also observed, but no damaging variants in this region have been defined by functional studies and no pathogenic variants have yet been observed in the clinically tested population. According to the BRCA1-NVM model, the total proportion of all theoretically possible damaging variants in BRCA1 is ~8%, almost all of which are located in the known RING and BRCT domains. For BRCA2, family-based studies in combination with the Align-GVGD prediction method were previously used to estimate that 33% of missense variants in the BRCA2 DNA binding domain were damaging.35 While based on small numbers of missense variants, this is consistent with predictions from the NVM and RF models for BRCA2, although the frequency based on the BRCA2-NVM and BRCA2-RF models is as high as 50% in specific regions. A notable drop in the estimated pathogenic potential was observed in the BRCA2 OB2 DNA binding domain. This was also observed when considering all pathogenic BRCA2 missense pathogenic variations listed in ClinVar.
However, it should be noted that when applied to genes other than BRCA1 and BRCA2 (Table S6) (or even BRCA1-RF or -NVM and applied to BRCA2 and vice versa), the performance of the NVM and RF models was much lower, with MCCs <0.40, as shown for the BRCA2-NVM (Table S7). The sizable reduction in model accuracy suggested that recalibrated models are specific to the initial gene of interest and cannot be effectively extrapolated to other disease genes. Another potential explanation for this phenomenon is that not all missense variants in other genes may exert phenotypic effects through loss of activity. Because the BRCA1 and BRCA2 assays are limited to measurement of loss of function, perhaps more comprehensive assays to evaluate splicing alterations, gain-of-function pathogenic variations, and epigenetic influences on gene function are needed to extend the NVM and RF prediction models to other genes. Separately, disruption of functions other than transcriptional activation or homology directed repair by missense variants could result in recalibration of the NVM and RF prediction models. Other influences on model performance may include AT versus GC content of coding sequences and codon usage, and the structural effects of observed variants. Finally, the differences could be due to evolutionary constraint because some models like Align GVGD and PolyPhen2 perform well for the highly conserved BRCA1, but profoundly less so for the less constrained BRCA2.
While the clinical implications of truncating pathogenic variations in the BRCA1 and BRCA2 breast cancer predisposition genes are clear, interpretation of missense variants is more challenging. Here we present an approach for predicting the functional impact and potentially the pathogenicity of missense BRCA1 and BRCA2 variants, based on functional evaluation of variants and in silico sequence-based analysis. The functional studies of BRCA2 variants in combination with similar studies of BRCA1 now identify 130 variants in these genes that are damaging and likely pathogenic and may substantially increase risk of breast, ovarian, and other cancers. In contrast, public databases currently identify fewer than 40 such variants. In the absence of functional results, other methods for variant assessment are needed. Many in silico prediction methods exist for characterization of missense variants, but the interpretation of results from these methods, and the accuracy of the methods for predicting whether variants in BRCA1 and BRCA2 are damaging or neutral, are not well defined. Here we recalibrated established in silico prediction methods for missense variants using results from BRCA1 and BRCA2 functional assays and developed RF and NVM models that incorporate multiple in silico prediction methods. These classifiers outperformed the individual in silico models. Overall this approach leverages measures of BRCA1 and BRCA2 functional activity to improve the classification of BRCA1 and BRCA2 VUS detected by clinical genetic testing and tumor sequencing.
References
Easton DF. How many more breast cancer predisposition genes are there? Breast Cancer Res. 1999;1:14–17.
Campeau PM, Foulkes WD, Tischkowitz MD. Hereditary breast cancer: new genetic developments, new therapeutic avenues. Hum Genet. 2008;124:31–42.
Pal T, Permuth-Wey J, Betts JA, et al. BRCA1 and BRCA2 mutations account for a large proportion of ovarian carcinoma cases. Cancer. 2005;104:2807–16.
Landrum MJ, Lee JM, Benson M, et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2016;44:D862–868. D1
Eggington JM, Bowles KR, Moyes K, et al. A comprehensive laboratory-based program for classification of variants of uncertain significance in hereditary cancer genes. Clin Genet. 2014;86:229–37.
Guidugli L, Pankratz VS, Singh N, et al. A classification model for BRCA2 DNA binding domain missense variants based on homology-directed repair activity. Cancer Res. 2013;73:265–75.
Millot GA, Carvalho MA, Caputo SM, et al. A guide for functional analysis of BRCA1 variants of uncertain significance. Hum Mutat. 2012;33:1526–37.
Iversen ES Jr, Couch FJ, Goldgar DE, et al. A computational method to classify variants of uncertain significance using functional assay data with application to BRCA1. Cancer Epidemiol Biomark Prev. 2011;20:1078–88.
Ng PC, Henikoff S. SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31:3812–4.
Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–9.
Cooper GM, Stone EA, Asimenos G, et al. Distribution and intensity of constraint in mammalian genomic sequence. Genome Res. 2005;15:901–13.
Tavtigian SV, Byrnes GB, Goldgar DE, et al. Classification of rare missense substitutions, using risk surfaces, with genetic- and molecular-epidemiology applications. Hum Mutat. 2008;29:1342–54.
Kircher M, Witten DM, Jain P, et al. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46:310–5.
Stenson PD, Ball EV, Mort M, et al. The Human Gene Mutation Database (HGMD) and its exploitation in the fields of personalized genomics and molecular evolution. Curr Protoc Bioinformatics. 2012;1:113.
Starita LM, Young DL, Islam M, et al. Massively parallel functional analysis of BRCA1 RING domain variants. Genetics. 2015;200:413–22.
Woods NT, Baskin R, Golubeva V, et al. Functional assays provide a robust tool for the clinical annotation of genetic variants of uncertain significance. NPJ Genom Med. 2016;1:16001.
Lee MS, Green R, Marsillac SM, et al. Comprehensive analysis of missense variations in the BRCT domain of BRCA1 by structural and functional assays. Cancer Res. 2010;70:4880–90.
Farrugia DJ, Agarwal MK, Pankratz VS, et al. Functional assays for classification of BRCA2 variants of uncertain significance. Cancer Res. 2008;68:3523–31.
Lindor NM, Guidugli L, Wang X, et al. A review of a multifactorial probability-based model for classification of BRCA1 and BRCA2 variants of uncertain significance (VUS). Hum Mutat. 2012;33:8–21.
Goldgar DE, Easton DF, Deffenbaugh AM, et al. Integrated evaluation of DNA sequence variants of unknown clinical significance: application to BRCA1 and BRCA2. Am J Hum Genet. 2004;75:535–44.
Liu X, Wu C, Li C, et al. dbNSFPv3.0: a one-stop database of functional predictions and annotations for human nonsynonymous and splice-site SNVs. Hum Mutat. 2016;37:235–41.
Kocher JP, Quest DJ, Duffy P, et al. The Biological Reference Repository (BioR): a rapid and flexible system for genomics annotation. Bioinformatics. 2014;30:1920–2.
McLaren W, Pritchard B, Rios D, et al. Deriving the consequences of genomic variants with the Ensembl API and SNP Effect Predictor. Bioinformatics. 2010;26:2069–70.
Matthews BW. Comparison of the predicted and observed secondary structure of T4 phage lysozyme. Biochim Biophys Acta. 1975;405:442–51.
Lopez-Raton M, Cadarso-Suarez C, Rodriguez-Alvarez MX, et al. OptimalCutpoints: an R package for selecting optimal cutpoints in diagnostic tests. J Stat Softw. 2014;61:1–36.
Liaw A, Wiener M. Classification and regression by randomForest. R News. 2002;2/3:18–22.
Feng B, Goldgar D. An integrated framework for sequence variant prioritization. American Society of Human Genetics Annual Meeting. 2014;Abstract 1370T.
Carter H, Douville C, Stenson PD, et al. Identifying Mendelian disease genes with the variant effect scoring tool. BMC Genom. 2013;14 Suppl 3:S3.
Lopes MC, Joyce C, Ritchie GR, et al. A combined functional annotation score for non-synonymous variants. Hum Hered. 2012;73:47–51.
Gonzalez-Perez A, Lopez-Bigas N. Improving the assessment of the outcome of nonsynonymous SNVs with a consensus deleteriousness score, Condel. Am J Hum Genet. 2011;88:440–9.
Wang Q, Zhang H, Kajino K, et al. BRCA1 binds c-Myc and inhibits its transcriptional and transforming activity in cells. Oncogene. 1998;17:1939–48.
Zhong Q, Chen CF, Li S, et al. Association of BRCA1 with the hRad50-hMre11-p95 complex and the DNA damage response. Science. 1999;285:747–50.
Xia B, Sheng Q, Nakanishi K, et al. Control of BRCA2 cellular and clinical functions by a nuclear partner, PALB2. Mol Cell. 2006;22:719–29.
Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24.
Easton DF, Deffenbaugh AM, Pruss D, et al. A systematic genetic assessment of 1,433 sequence variants of unknown clinical significance in the BRCA1 and BRCA2 breast cancer-predisposition genes. Am J Hum Genet. 2007;81:873–83.
Guidugli L, Shimelis H, Masica DL, et al. Assessment of the Clinical Relevance of BRCA2 Missense Variants by Functional and Computational Approaches. Am J Hum Genet. E-pub ahead of print 17 January 2018.
Funding
This work was supported by the Breast Cancer Research Foundation; National Institutes of Health (grants CA192393, CA176785, CA116167); and a National Cancer Institute Specialized Program of Research Excellence (SPORE) grant in Breast Cancer to Mayo Clinic (P50 CA116201).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Disclosure
The authors declare no conflicts of interest.
Rights and permissions
About this article

Cite this article
Hart, S.N., Hoskin, T., Shimelis, H. et al. Comprehensive annotation of BRCA1 and BRCA2 missense variants by functionally validated sequence-based computational prediction models. Genet Med 21, 71–80 (2019). https://doi.org/10.1038/s41436-018-0018-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-018-0018-4
Keywords
This article is cited by
-
Analyzing the effects of BRCA1/2 variants on mRNA splicing by minigene assay
Journal of Human Genetics (2023)
-
Understanding and predicting the functional consequences of missense mutations in BRCA1 and BRCA2
Scientific Reports (2022)
-
An integrative model for the comprehensive classification of BRCA1 and BRCA2 variants of uncertain clinical significance
npj Genomic Medicine (2022)
-
Saturation variant interpretation using CRISPR prime editing
Nature Biotechnology (2022)
-
Breast cancer risks associated with missense variants in breast cancer susceptibility genes
Genome Medicine (2022)
