
Abstract
Resistance to M. tuberculosis, often referred to as “RSTR” in the literature, is being increasingly studied because of its potential relevance as a clinical outcome in vaccine studies. This review starts by addressing the importance of epidemiological characterization of this phenotype, and ongoing challenges in that characterization. Then, this review summarizes the extant genetic and genomic studies of this phenotype, including heritability studies, candidate gene studies, and genome-wide association studies, as well as whole transcriptome studies. Findings from recent studies that used longitudinal characterization of the RSTR phenotype are compared to those using a cross-sectional definition, and the challenges of using tuberculin skin test and interferon-gamma release assay are discussed. Finally, future directions are proposed. Since this is a rapidly evolving area of public health significance, this review will help frame future research questions and study designs.
Similar content being viewed by others

Introduction
Tuberculosis (TB) remains a major public health burden globally, with roughly one-quarter to one-third of the world infected by the disease-causing bacterium Mycobacterium tuberculosis (Mtb). It remains a critical public health issue because it one of the top infectious disease causes of mortality worldwide, in recent years falling a close second only to COVID-19 in mortality [1]. While it its public health significance is great, there is still a great need for improved treatment and vaccination strategies [2], making the identification of susceptibility factors a major focus of basic science research.
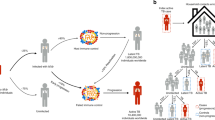
After exposure to an infectious tuberculosis (TB) case, there are three possible outcomes: resistance or early clearance of the bacillus, asymptomatic or latent Mtb infection (LTBI) that can persist for decades, or symptomatic ‘active TB’, which includes pulmonary disease that can result in further transmission. Active TB is manifested typically through a severe productive cough (pulmonary TB), though it can also affect other organ systems (extrapulmonary TB), and it is also commonly characterized by fever, weight loss, night sweats, and other clinical characteristics common to respiratory infectious diseases. Resistance to infection is seen in individuals who are initially uninfected based on standard diagnostic tests such as the tuberculin skin test (TST) and/or interferon-gamma release assay (IGRA), and then remain negative. Some individuals may test negative initially, but within months, or perhaps even longer, convert to an LTBI state (Fig. 1). While both the uninfected state and LTBI state are asymptomatic, LTBI individuals have a 10% lifetime risk of progression to active symptomatic TB.

After initial exposure to an active TB case, ascertained individuals may present as either infected or uninfected based on the tuberculin skin test (TST) and/or interferon-gamma release assay (IGRA) such as the QuantiFERON. After this initial exposure, some individuals remain uninfected, while others convert to positive. This conversion can happen weeks or even months after that initial exposure.
While previous TB research focused primarily on factors associated with the transition from LTBI to active TB disease, recent attention has turned to factors associated with resistance to developing LTBI after exposure, as it would provide an appealing target for prevention, specifically through vaccine development and host-directed therapies [3]. The underlying premise is that targeting this stage of earlier pathogenesis, the progression from the uninfected to the infected state, might prevent LTBI and ultimately progression to disease [4]. This is similar in principle to the identification of biologic mechanisms that prevent HIV infection (as opposed to development of full-blown disease) that has been harnessed as a treatment modality [5]. A vaccine that prevents infection would be easier to study in clinical trials, since infection events are more common than disease events, and indeed, such studies are ongoing [4]. Epidemiologically, this resistance phenotype has also gained attention in understanding annual risk of infection [6]. Accurate estimates of the annual risk of infection are important for understanding the global burden of disease and for developing public health strategies to slow TB transmission. Understanding LTBI, and in converse resistance to LTBI, is critical for understanding the complexity of immune responses in TB pathogenesis [3, 7]. Genetics and genomics provide one perspective into innate biologic factors that influence resistance, and thus are one avenue into target discovery, just like the original discovery of the CCR5 HIV-protective variant.
Identification of epidemiological factors associated with this ‘resistance’ phenotype has been challenging because of the wide variety of phenotype definitions used across studies [8], described in more detail below. Multiple studies have not identified clear epidemiologic factors that differentiated resisters from LTBIs [9,10,11,12]. A multinational study identified relationship to the index case and alcohol use as potential factors associated with resistance to infection, though these were no longer significant when considered in a multivariable model [13]. One study found that BCG vaccination was associated with resistance [14], though this study used a different clinical definition and it is not clear how this might have affected that finding (see further discussion below). Age might be an important factor since it is associated with time to TST conversion [15] as well as the aforementioned potential effect of BCG [14], though the other studies did not replicate this. Age as a potential confounder is noteworthy since it has also been proposed as a potential confounder of TB (disease) genetics studies [16]. While time spent on anti-retroviral treatment might be a potential factor [12], another study found no association between HIV infection (regardless of treatment) and resistance [17], so the importance of HIV infection itself is not well understood.
The ‘resister’ phenotype is also becoming the focus of genetic and genomic studies. In fact, the first study of this phenotype was actually a genetic study [18], and it was conducted prior to detailed epidemiological investigations. When designing and interpreting these studies, the definition of this phenotype is critical. In this review, first we focus on phenotype definition, then we turn our attention to current findings and potential future directions. While the focus of this paper is on tuberculosis, these principles are generally relevant to the genetic study of infectious diseases in general.
Resistance to infection: study design is critical
Individuals who despite prolonged exposure to infectious TB cases continue to have a persistently negative tuberculin skin test (TST) or interferon-γ response assay (IGRA) are of particular interest as they may help inform prevention strategies. Such individuals are presumed resistant to latent Mtb infection (RSTR) (Fig. 1). Controls (non-RSTR) would then be individuals who do not remain TST-/IGRA- after exposure. While this sounds simple, there are many elements of this definition that are critical and also sometimes challenging to implement. Epidemiological definitions in a variety of settings and immunological implications have been discussed at length in other reviews [3, 7, 8], so here we will briefly summarize (Table 1).
Exposure
In order to be considered a RSTR, exposure to an infectious TB case must be clearly established. As seen across the studies summarized in our review [8], different studies have approached this documentation of exposure differently. In close contact settings such as households, aspects of the TB diagnosis in the index case (culture confirmation, microbial load, etc.) are important, since these variables reflect potential infectiousness of that index case. In addition, data on proximity to the index TB case are also important, as they may additionally reflect risk of high intensity exposure. By contrast, in high transmission community-based settings, there is no index case. However, a community-based study in South Africa, a hyper-endemic setting, stated that transmission occurs more frequently in the community than in the household, and this transmission was verified based on positive antibody titers in TST-/IGRA- subjects [12]. A high degree of exposure may also result from certain employment settings, especially where TB is endemic [11]. However, no design is perfect. The household contact design does not guarantee transmission [19], and in populations of low endemicity, it is not clear that a community cohort would have sufficient exposure to truly characterize TST-/IGRA- individuals as “resistant” to infection.
Another proxy for exposure is age. As described above, some studies suggest that age might be an important risk factor for TST/IGRA conversion. Many of the other studies where age was not found to be a factor only included adults. Thus, age might be an appropriate variable to consider as an indicator of lifetime exposure. A recent simulation study has suggested that misclassification of exposure in cases and controls can bias genetic association study results [20, 21], so documentation of exposure is not a trivial point.
Duration of follow-up
The duration of follow-up in studies of “resisters” or “early clearance” has varied across studies. Cross-sectional studies are insufficient; exposure to a TB case might not translate into a positive test (IGRA and/or TST) in the contact until a few weeks later. Some studies have limited follow-up to 3 months after exposure [14], which is when potentially most conversions to positive TST/IGRA occur. However, our studies [15, 22] show that conversion to a positive test can occur 6 months or more from initial diagnosis of the TB case. Other studies have used 12 [10, 12] or 24 [15] months of follow-up. Our long term follow-up study [17] shows that conversion happens even several years (with an average length of follow-up of 9–10 years) after that initial exposure. In that case, acquisition of infection is likely due to exposure to some other TB case other than the original index, which may be more similar to the high endemicity situation described above. In either case, the point remains: cross-sectional studies and/or short-term follow-up (less than 6 months) might be insufficient for a strict RSTR definition. Lastly, our studies [17] and others [12] have shown that IGRA results collected over the course of a year may be unstable and clinical classifications are best made using the entire collection of data (IGRA and TST if available) over the course of observation for each subject. If only one or even two measurements were available, a subject might be misclassified due to a sporadic value.
Diagnostic tool
Early studies of the RSTR phenotype only used the TST, because the IGRA hadn’t been developed or refined yet. Thus, many published genetic studies are based on that phenotype. More recent studies are transitioning to the IGRA. This raises a new question, since discordance between these two tests is well-known [10, 17]. Will studies that use only the IGRA appropriately reflect ‘resistance’? Should studies use both tests, which might be logistically challenging, especially for longitudinal study designs? Ongoing studies need to tackle this question both epidemiologically and immunologically, to understand how different clinical definitions might be reflected biologically.
Lingering questions
The discussion above already mentions some key challenges in defining the RSTR phenotype. The first question is, how long must one conduct follow-up in a cohort study setting in order to be sufficiently certain about the characterization of RSTRs? In Stein et al. [17], ~80% of persistently TST- individuals remained both TST- and were also IGRA- after an average of 9-10 years post initial exposure. While that is a high concordance rate, is that high enough? Certainly it is logistically challenging and expensive to carry out follow-up studies for a decade or more. Based on their summary of the literature, Gutierrez et al. [8] advocate for at least 1 year of follow-up, since that is the period covering the majority of TST/IGRA conversions post-exposure. Kroon et al. [12] additionally advocate for the recruitment of especially high risk individuals, particularly HIV-infected, who are more likely to convert their TST/IGRA in a shorter amount of time. It is striking that antibody signatures identified in HIV-infected resisters with shorter follow-up are very similar to those found in the Ugandan long-term follow-up study [23], suggesting that at least this aspect of biological mechanism is robust to duration of follow-up as well as exposure (household vs community). It is also quite possible that additional genomic studies, building on those described below, may elucidate the importance of phenotypic stringency on biological interpretation. Additional long-term follow-up studies that quantify the conversion rate as a function of time since initial exposure (in household contact studies) or initial ascertainment (for community-based studies) are needed to better understand this issue.
Second, the question about TST/IGRA discordance and the field transitioning to the IGRA is not trivial. The objections to the TST have been based primarily on concerns of false positives due to BCG vaccination and/or boosting. However, many studies have not found a bias in TST conversion rates due to history of BCG vaccination, and studies of boosting suggest this effect is minimal [24].
Third, while above we outline variables that should be used in defining sufficient exposure, little work has been done examining the degree of exposure and whether this has an impact. While the Ugandan studies examined a quantitative epidemiologic risk score to define degree of exposure [17, 25], another large multinational study categorized exposure into low, medium, and high levels [13]; perhaps these quantitative levels would be helpful for future genetic analyses.
How is TB unique?
From a genetic epidemiological standpoint, it is important to consider which of these aspects of study design and phenotype definition are also relevant to the genetic study of other infectious diseases. The first point to consider is mode of transmission. TB is spread through airborne droplets, which is an indirect mode of transmission. This is one of the reasons why quantification of exposure is challenging, because it is not a direct mode of transmission as is seen in sexually transmitted diseases and other viral infections. Certainly establishing exposure for those infections is also challenging but entails a different strategy, which is beyond the scope of this review. Other infectious diseases with indirect transmission might be vector-borne or vehicle-borne, where establishing exposure is likely even more difficult, though as pointed out by the aforementioned simulation study [20], still important. The prevalence of infection and disease might also be a consideration when considering exposure. Second, not all infectious diseases have a stage of pathogenesis where the pathogen is present but does cause symptoms. Influenza and COVID-19 are examples of viral infectious where there is an asymptomatic state, and resistance in hepatitis C has been examined in terms of “spontaneous clearance” [20] (similar to the “early clearance” term coined in the Mtb resistance field [14]). In this same vein, different types of pathogens (bacteria, viruses, parasites) have different stages of pathogenesis, which ultimately could affect clinical definitions in genetic studies.
Recent genetic and genomic findings
Heritability – impact on gene mapping
A recent review by Abel et al. [16] nicely summarizes heritability estimates across a number of studies, with a variety of phenotype definitions and study designs, and in a variety of global populations. These values ranged from as low as 39% (for IGRA results) to 92% (for quantitative TST result). In our recent study, where we defined RSTR based on our long-term follow-up study [17], we found an estimate of 48% when including both HIV-infected and HIV-uninfected individuals in the analysis. In a sense, our RSTR definition is a composite phenotype, requiring negative results on both TST and IGRA, so not directly comparable to previous studies, but it also has longitudinal stringency. The heritability analysis of other TB phenotypes presented in McHenry et al. illustrates that strictness of phenotype yields higher heritability estimates. As a result, higher heritability estimates result in higher power for gene mapping. This is important for interpretation of the extant literature.
Candidate gene and genome-wide association studies (GWAS)
Most of the initial studies examining LTBI (or its converse, lack of infection) were based on cross-sectional studies, sometimes in low transmission settings (Table 2). Other studies examined TST longitudinally, but did not use IGRA in characterization of infected and uninfected individuals because that assay was unavailable at the time. Only recently as IGRAs became a standard part of epidemiological studies have genetic studies also characterized subjects according to this measure. Studies conducted early on were primarily focused on candidate genes and genome-wide linkage studies, and more recently as genotyping costs have come down considerably, genome-wide association studies have been conducted. This contrast in approach makes comparison across papers difficult, as candidate gene studies required less stringent p-values for declaring significance, and linkage studies also require a lower significance threshold than GWAS. Linkage studies also have low resolution for identifying common variants. The passage of time has affected the available phenotyping strategies in addition to the genotyping technology. If our goal is to use genetics to identify potential therapeutic targets, replication at the gene level is of primary interest. Frankly, this is the only way to consider replication between linkage results and association studies, since linkage studies mostly employ allele-sharing between relative pairs as the statistic of interest, and as such do not identify an effect allele. In addition, targeted candidate gene studies might employ different strategies for selecting single nucleotide polymorphism (SNP) markers than GWAS studies.
A few observations can be made based on the results of the literature summarized in Table 2. This table includes the primary findings of the cited studies, and also indicates which of these findings were replicated (at p < 0.05) in a recent GWAS [26] where the phenotype was based on stringent follow-up and both TST and IGRA [17]. Thus, this enables examination of replication by phenotype definition. First, there was one locus replicated between original linkage studies using TST only, and that was on 5p15. This locus appears to be robust because it was observed in two different populations (South Africa and Uganda), using slightly different TST-based definitions (persistently negative TST and continuous value of TST induration), and most recently replicated in a GWAS that used the stringent definition of TST/IGRA persistent negativity. Second, some of the loci originally identified by genome-wide linkage studies (2q21-q24 and 11p14), again using different variations of the TST-based definition (longitudinal and cross-sectional, respectively), were also replicated by the recent GWAS that used the stringent TST/IGRA definition. Third, the GWAS studies uncovered significant loci that were not identified by the genome-wide linkage studies. This is quite likely due to the increased power and resolution of association analysis methodology. It is also possible that the more stringent definitions yielded increased statistical power [26]. However, with only two such studies, this still provides justification for additional GWAS studies. The consistency between the older TST-based linkage and association studies and McHenry et al. [26] suggests two things: One, the high heritability also identified by McHenry et al. [26] suggests that this strict phenotype may have increased power to detect effects. Two, since these loci were originally identified with a variety of phenotype definitions but still attained genome-wide significance, perhaps effects seen at this stringent a threshold may be robust to phenotype definition.
The non-replication between the McHenry et al. [26] and Quistrebert et al. [27] studies is curious and deserves particular attention. The Quistrebert et al. [27] study was interesting in that it used 3 different populations for internal replication, with rigorous criteria for TST and IGRA positivity, but two of the three cohorts only utilized TST and IGRA at baseline with no longitudinal assessment. The lack of replication of findings between the two GWAS studies reveals that the impact of a stringent phenotype definition might increase power to detect effects, but that does not guarantee replication, though this lack of replication may also suggest genetic heterogeneity underlying RSTR. It is also unclear whether the lack of replication between the two GWAS studies was due to difference in phenotypic characterization (cross-sectional vs long-term follow-up) or perhaps ethnic background. Certainly, the use of 3 ethnically diverse populations within the Quistrebert study is a major strength. Since the study focused on the loci that were internally replicated, it is unknown if there might be replication with some of the other loci listed in Table 2. Furthermore, it is quite possible that there are additional as-yet undetected genetic associations with the RSTR phenotype. Indeed, the analysis by Dawkins et al. [28] revealed genetic associations with HLA that had not been seen in prior studies, and the authors hypothesized that these associations were detected because of the well-defined phenotypic contrasts.
The validity of these genetic associations is also difficult to assess for a few reasons. First, as is very clear from the citation list, relatively few study groups have conducted large scale studies of this phenotype, therefore there are relatively few opportunities for replication. This was a stated weakness of the Ugandan GWAS – the analysis only contained a discovery sample with no replication sample. Second, the functional implications of these loci have not been delineated. If the goal is to use such findings to identify therapeutic targets, this is an important next step. Most likely, some of these studies have been attempted and/or are ongoing, so it will be essential to keep a close eye on the literature. Intriguingly, the loci identified by these studies are distinct from those associated with TB susceptibility [29,30,31], suggesting that the biologic underpinnings of resistance are quite different.
Transcriptomic studies
As of this writing, only a few RNA expression studies have been published, though there are likely many more ongoing (Table 3). While there are only a few studies, they also include functional validation, which is an important step towards the ultimate goal of identifying biologic targets. The first study used microarray technology, which was cutting-edge at the time, and the phenotype was based on TST only, because again, QFT was not used at the time [32]. This study examined macrophage gene expression after stimulation with Mtb in vitro. Several gene sets were differentially expressed, but the most notable pathway was the histone deacetylase pathway, and these findings were validated with immunologic and microbiologic experiments.
The second two studies utilized RNA-seq technology and the stricter RSTR phenotype [17] described above; the first of these examining basal gene expression levels in macrophages [33], and the second examining expression responses after these macrophages were stimulated with Mtb in vitro [34], similar to the aforementioned microarray study. The study examining basal gene expression levels also included two populations (Ugandan RSTRs and South African miners with the RSTR phenotype), and identified gene sets that were associated with carbon metabolism and free fatty acid enrichment that differentiated RSTRs and LTBI in both populations. These hypotheses were validated with a variety of functional validation experiments as well as candidate gene association analysis using SNP data. In the study where gene expression after stimulation with Mtb in vitro was examined, again no single gene differences were identified after multiple testing correction, but gene set and pathway enrichment analysis revealed several pathways of interest, most notably TNF. Validation was conducted again using SNP association analysis, and these analyses revealed associations with ABCA1 and DUSP2. Note that the RNA-seq study [34] did not replicate the microarray study [32]; it is unknown whether this lack of replication was due to small sample size, different phenotype definition, or different technology. These questions can only be elucidated with additional studies and functional validation.
As of the writing of this review, all three published transcriptomic studies have been based in Uganda (with one replicating findings in South Africa), and they have differed by technology, phenotype, and tissue of interest (stimulated or unstimulated macrophages). Thus, it is impossible to generalize these findings to other populations, either those with different phenotype definitions or those with different exposure profiles. This will certainly be a focus of future research. However, it is worth noting that the most novel findings from these current transcriptomic studies are different from those identified with TB disease [35,36,37,38,39,40].
Future directions
While there are no published studies as yet examining epigenetic influences on the RSTR phenotypes, there is another clinical framework that provides premise for these studies. Progression, or non-progression, to TB disease, in the setting of HIV infection is another model for resistance. When immunosuppressed individuals living in TB-endemic settings do not develop TB disease or LTBI despite exposure and high risk, this provides a model of resistance that is useful for gene mapping [41, 42]. Two studies under this paradigm have revealed important epigenetic effects underlying resistance to infection. First, a methylome-wide study, examining methylation markers across the genome, identified methylation markers associated with TB susceptibility as well as SNP markers that interacted with methylation markers to increase susceptibility for TB [43]. Second, a study examining chromatin accessibility using ATAC-seq identified differential chromatin accessibility in different clinical groups [44]. Certainly future studies of the RSTR phenotype will utilize these technologies to better understand the impact of epigenetics in the context of genetic and transcriptomic variation. In addition, eQTL studies may begin to integrate SNP association and transcriptomic data, which will enable interpretation of the role of both types of variation in clinical outcome [45]. The integration of SNP and RNA data will provide insight into function, which is essential for developing therapeutic strategies.
In addition, the lack of functional validation of findings from genetic studies illustrate the challenge in going from an association (or linkage) effect to a biologic readout. One approach that shows promise is using genetic data from a GWAS to validate transcriptomic findings, as in two of our studies [33, 34]. Another approach is to utilize genetic variation data within a bioinformatic approach that integrates data generated across other ‘omic platforms. Analyses that examine genetic variants one by one overlook possible epistatic and epigenetic effects, a point mentioned by another recent review in this area [16]. Systems biology approaches that incorporate genetic and genomic data along with other ‘omic (proteomic, epigenetic, etc) data may reveal interactions between these various components of a comprehensive biologic model [46,47,48,49,50,51,52,53,54,55,56]. Yet another related approach that may point to disease biology is to examine enriched gene sets and pathways [30]. This has proven fruitful in both our GWAS [26] and transcriptomic studies [32,33,34], especially because the gene expression studies did not reveal any single gene effects that were significant after multiple testing correction. Ultimately, genomic data are one piece in the puzzle, and should be integrated with the findings from immunologic and ‘omic strategies.
While replication analyses have been conducted independently, another future goal would be a cross-population meta-analysis. The challenge here would be accounting for variability in phenotype definition. Given some of the replication seen between some of the studies using cross-sectionally defined phenotypes and longitudinally-defined phenotypes, it would be interesting to see if GWAS using cross-sectional phenotypes may add some insight to the question about robustness of genetic results in the context of phenotype definition. A recent meta-analysis of TB disease GWAS studies was conducted [57], and it illustrated the challenges of phenotype definitions different by geographic location. Still, this goal is commendable because it may bring to the fore genetic associations that did not attain significance after multiple testing correction in samples independently, but at lower yet suggestive levels, these loci may still be clinically and biologically meaningful. Such results might not be extracted at lower levels of significance unless they were combined with other samples. However, for a meta-analysis to be valuable, first, genomic studies of the RSTR phenotype need to be conducted more diverse populations. As noted above, the currently published findings come from a few well-characterized but globally restricted populations. For a biologic target to have any translatability, it needs to be transferable across global populations. It is also worth re-analyzing published TB GWAS data (where disease was the focus, not resistance to infection) to examine whether genes associated with resistance might also be associated with disease progression at lower levels of significance and/or different directions of effect. Lastly, it is of interest to understand if the regions associated with resistance have arisen due to selective pressure. Signatures of selection have been observed in regions previously associated with TB disease resistance in the context of HIV infection [42]. Addressing this question would entail the analysis and collection of new data, and is beyond the scope of this review, but a worthy question for future studies.
Conclusion
Genetic and genomic studies of resistance to Mtb infection are increasingly important as they become part of multidisciplinary to identify potential vaccine and other therapeutic targets. Relatively few studies have examined this strictly defined phenotype, though recent work suggests that genetic findings may be potentially robust to phenotype definition. Future efforts must attempt to use similarly stringent phenotype definitions in order to determine the true impact on reliability and validity of genetic and transcriptomic associations. In addition, studies are needed in a variety of global settings in order to better understand the impact of Mtb genetic variation and other environmental factors the reliability and validity of these genetic associations. These next steps are critical when considering potential translation of these findings to at-risk human populations. Moreover, it is important to understand how these results compare to those currently being harnessed as host-directed therapy and vaccine targets for preventing disease progression as opposed to maintaining resistance to infection. However, before the findings from these studies are translatable, there must be consideration of how such prevention strategies fit in the scope of existing strategies to prevent progression to TB disease. The public health strategies may differ depending on the biologic commonalities from the results of ongoing and future studies, and this will impact how potential host-directed therapies and/or novel vaccine approaches are implemented.
Data availability
Data sharing not applicable to this article as no datasets were generated or analysed during the current study.
References
World Health Organization. WHO Tuberculosis fact sheet, 2023.
Uplekar M, Weil D, Lonnroth K, Jaramillo E, Lienhardt C, Dias HM, et al. WHO’s new end TB strategy. Lancet. 2015;385:1799–801. https://doi.org/10.1016/s0140-6736(15)60570-0.
Simmons JD, Stein CM, Seshadri C, Campo M, Alter G, Fortune S, et al. Immunological mechanisms of human resistance to persistent Mycobacterium tuberculosis infection. Nat Rev Immunol. 2018;18:575–89. https://doi.org/10.1038/s41577-018-0025-3.
Garcia-Basteiro, AL, White, RG, Tait, D, Schmidt, AC, Rangaka, MX, Quaife, M et al. End-point definition and trial design to advance tuberculosis vaccine development. European respiratory review: an official journal of the European Respiratory Society. 2022;31, https://doi.org/10.1183/16000617.0044-2022.
Gulick RM, Lalezari J, Goodrich J, Clumeck N, DeJesus E, Horban A, et al. Maraviroc for previously treated patients with R5 HIV-1 infection. N Engl J Med. 2008;359:1429–41. https://doi.org/10.1056/NEJMoa0803152.
Dowdy DW, Behr MA. Are we underestimating the annual risk of infection with Mycobacterium tuberculosis in high-burden settings? Lancet Infect Dis. 2022;22:e271–8. https://doi.org/10.1016/s1473-3099(22)00153-0.
Boom, WH, Schaible, UE & Achkar, JM. The knowns and unknowns of latent Mycobacterium tuberculosis infection. J Clin Invest. 2021;131, https://doi.org/10.1172/jci136222.
Gutierrez J, Kroon EE, Möller M, Stein CM. Phenotype definition for “resisters” to mycobacterium tuberculosis infection in the literature-a review and recommendations. Front Immunol. 2021;12:619988 https://doi.org/10.3389/fimmu.2021.619988.
Stein CM, Nsereko M, Malone LL, Okware B, Kisingo H, Nalukwago S, et al. Long-term stability of resistance to latent mycobacterium tuberculosis infection in highly exposed tuberculosis household contacts in Kampala, Uganda. Clin Infect Dis. 2019;68:1705–12. https://doi.org/10.1093/cid/ciy751.
Mave V, Chandrasekaran P, Chavan A, Shivakumar S, Danasekaran K, Paradkar M, et al. Infection free “resisters” among household contacts of adult pulmonary tuberculosis. PLoS One. 2019;14:e0218034 https://doi.org/10.1371/journal.pone.0218034.
Chihota VN, Ntshiqa T, Maenetje P, Mansukhani R, Velen K, Hawn TR, et al. Resistance to Mycobacterium tuberculosis infection among highly TB exposed South African gold miners. PLoS One. 2022;17:e0265036 https://doi.org/10.1371/journal.pone.0265036.
Kroon EE, Kinnear CJ, Orlova M, Fischinger S, Shin S, Boolay S, et al. An observational study identifying highly tuberculosis-exposed, HIV-1-positive but persistently TB, tuberculin and IGRA negative persons with M. tuberculosis specific antibodies in Cape Town, South Africa. EBioMedicine. 2020;61:103053 https://doi.org/10.1016/j.ebiom.2020.103053.
Baliashvili D, Gandhi NR, Kim S, Hughes M, Mave V, Mendoza-Ticona A, et al. Resistance to mycobacterium tuberculosis infection among household contacts: a multinational study. Clin Infect Dis. 2021;73:1037–45. https://doi.org/10.1093/cid/ciab269.
Verrall, AJ, Alisjahbana, B, Apriani, L, Novianty, N, Nurani, AC, van Laarhoven, A et al. Early clearance of mycobacterium tuberculosis: the INFECT case contact cohort study in Indonesia. J Infect Dis. 2019; https://doi.org/10.1093/infdis/jiz168.
Stein CM, Zalwango S, Malone LL, Thiel B, Mupere E, Nsereko M, et al. Resistance and susceptibility to Mycobacterium tuberculosis infection and disease in tuberculosis households in Kampala, Uganda. Am J Epidemiol. 2018;187:1477–89. https://doi.org/10.1093/aje/kwx380.
Abel L, Fellay J, Haas DW, Schurr E, Srikrishna G, Urbanowski M, et al. Genetics of human susceptibility to active and latent tuberculosis: present knowledge and future perspectives. Lancet Infect Dis. 2018;18:e64–e75. https://doi.org/10.1016/s1473-3099(17)30623-0.
Stein CM, Nsereko M, Malone LL, Okware B, Kisingo H, Nalukwago S, et al. Long-term stability of resistance to latent M. tuberculosis infection in highly exposed TB household contacts in Kampala, Uganda. Clin Infect Dis. 2019;68:1705–12. https://doi.org/10.1093/cid/ciy751.
Stein CM, Zalwango S, Malone LL, Won S, Mayanja-Kizza H, Mugerwa RD, et al. Genome scan of M. tuberculosis infection and disease in Ugandans. PloS One. 2008;3:e4094.
Whalen CC, Zalwango S, Chiunda A, Malone L, Eisenach K, Joloba M, et al. Secondary attack rate of tuberculosis in urban households in Kampala, Uganda. PLoS One. 2011;6:e16137.
Duchen D, Vergara C, Thio CL, Kundu P, Chatterjee N, Thomas DL, et al. Pathogen exposure misclassification can bias association signals in GWAS of infectious diseases when using population-based common control subjects. Am J Hum Genet. 2023;110:336–48. https://doi.org/10.1016/j.ajhg.2022.12.013.
Duchen, D, Vergara, C, Thio, CL, Kundu, P, Chatterjee, N, Thomas, DL et al. Pathogen exposure misclassification can bias association signals in GWAS of infectious diseases when using population-based common controls. medRxiv, 2022.2007.2014.22276656, https://doi.org/10.1101/2022.07.14.22276656
Stein CM, Mayanja-Kizza H, Hawn TR, Boom WH. Importance of study design and phenotype definition in ongoing studies of resistance to latent mycobacterium tuberculosis infection. J Infect Dis. 2020;221:1025–6. https://doi.org/10.1093/infdis/jiz539.
Lu LL, Smith MT, Yu KKQ, Luedemann C, Suscovich TJ, Grace PS, et al. IFN-gamma-independent immune markers of Mycobacterium tuberculosis exposure. Nat Med. 2019;25:977–87. https://doi.org/10.1038/s41591-019-0441-3.
Sekandi JN, Zalwango S, Nkwata AK, Martinez L, Kakaire R, Mutanga JN, et al. Low prevalence of tuberculin skin test boosting among community residents in Uganda. Am J Trop Med Hyg. 2018;98:379–81. https://doi.org/10.4269/ajtmh.17-0591.
Ma N, Zalwango S, Malone LL, Nsereko M, Wampande EM, Thiel BA, et al. Clinical and epidemiological characteristics of individuals resistant to M. tuberculosis infection in a longitudinal TB household contact study in Kampala, Uganda. BMC Infect Dis. 2014;14:352.
McHenry ML, Benchek P, Malone L, Nsereko M, Mayanja-Kizza H, Boom WH, et al. Resistance to TST/IGRA conversion in Uganda: heritability and genome-wide association study. EBioMedicine. 2021;74:103727. https://doi.org/10.1016/j.ebiom.2021.103727.
Quistrebert J, Orlova M, Kerner G, Ton LT, Luong NT, Danh NT, et al. Genome-wide association study of resistance to Mycobacterium tuberculosis infection identifies a locus at 10q26.2 in three distinct populations. PLoS Genet. 2021;17:e1009392. https://doi.org/10.1371/journal.pgen.1009392
Dawkins BA, Garman L, Cejda N, Pezant N, Rasmussen A, Rybicki BA, et al. Novel HLA associations with outcomes of Mycobacterium tuberculosis exposure and sarcoidosis in individuals of African ancestry using nearest-neighbor feature selection. Genet Epidemiol. 2022;46:463–74. https://doi.org/10.1002/gepi.22490.
McHenry ML, Williams SM, Stein CM. Genetics and evolution of tuberculosis pathogenesis: New perspectives and approaches. Infect Genet evolution: J Mol Epidemiol Evolut Genet Infect Dis. 2020;81:104204. https://doi.org/10.1016/j.meegid.2020.104204.
Stein, CM, Sausville, L, Wejse, C, Sobota, RS, Zetola, NM, Hill, PC et al. Genomics of human pulmonary tuberculosis: from genes to pathways. Curr Genet Med Rep. 2017; https://doi.org/10.1007/s40142-017-0130-9.
Ndong Sima, CAA, Smith, D, Petersen, DC, Schurz, H, Uren, C & Möller, M. The immunogenetics of tuberculosis (TB) susceptibility. Immunogenetics. 2022; https://doi.org/10.1007/s00251-022-01290-5.
Seshadri C, Sedaghat N, Campo M, Peterson G, Wells RD, Olson GS, et al. Transcriptional networks are associated with resistance to Mycobacterium tuberculosis infection. PLoS One. 2017;12:e0175844 https://doi.org/10.1371/journal.pone.0175844.
Simmons, JDS, C M, Van, PT, Peterson, GJ, Grautsein, AD, Nguyen, FK, et al. In Pacific Tuberculosis Pathogenesis and Host-Response Symposium (Portland, Oregon, 2019).
Simmons, JD, Dill-McFarland, KA, Stein, CM, Van, PT, Chihota, V, Ntshiqa, T et al. Monocyte transcriptional responses to mycobacterium tuberculosis associate with resistance to tuberculin skin test and interferon gamma release assay conversion. mSphere, e0015922. 2022; https://doi.org/10.1128/msphere.00159-22.
Zak DE, Penn-Nicholson A, Scriba TJ, Thompson E, Suliman S, Amon LM, et al. A blood RNA signature for tuberculosis disease risk: a prospective cohort study. Lancet. 2016;387:2312–22. https://doi.org/10.1016/s0140-6736(15)01316-1.
Berry MP, Graham CM, McNab FW, Xu Z, Bloch SA, Oni T, et al. An interferon-inducible neutrophil-driven blood transcriptional signature in human tuberculosis. Nature. 2010;466:973–7. https://doi.org/10.1038/nature09247.
Anderson ST, Kaforou M, Brent AJ, Wright VJ, Banwell CM, Chagaluka G, et al. Diagnosis of childhood tuberculosis and host RNA expression in Africa. N. Engl J Med. 2014;370:1712–23. https://doi.org/10.1056/NEJMoa1303657.
Kaforou M, Wright VJ, Oni T, French N, Anderson ST, Bangani N, et al. Detection of tuberculosis in HIV-infected and -uninfected African adults using whole blood RNA expression signatures: a case-control study. PLoS Med. 2013;10:e1001538. https://doi.org/10.1371/journal.pmed.1001538.
Maertzdorf J, Repsilber D, Parida SK, Stanley K, Roberts T, Black G, et al. Human gene expression profiles of susceptibility and resistance in tuberculosis. Genes Immun. 2011;12:15–22.
Ottenhoff TH, Dass RH, Yang N, Zhang MM, Wong HE, Sahiratmadja E, et al. Genome-wide expression profiling identifies type 1 interferon response pathways in active tuberculosis. PLoS One. 2012;7:e45839. https://doi.org/10.1371/journal.pone.0045839.
Sobota RS, Stein CM, Kodaman N, Maro I, Wieland-Alter W, Igo RP Jr., et al. A chromosome 5q31.1 locus associates with tuberculin skin test reactivity in HIV-positive individuals from tuberculosis hyper-endemic regions in east Africa. PLoS Genet. 2017;13:e1006710. https://doi.org/10.1371/journal.pgen.1006710.
Sobota RS, Stein CM, Kodaman N, Scheinfeldt LB, Maro I, Wieland-Alter W, et al. A locus at 5q33.3 confers resistance to tuberculosis in highly susceptible individuals. Am J Hum Genet. 2016;98:514–24.
Stein CM, Benchek P, Bartlett J, Igo RP, Sobota RS, Chervenak K, et al. Methylome-wide analysis reveals epigenetic marks associated with resistance to tuberculosis in human immunodeficiency virus-infected individuals from East Africa. J Infect Dis. 2021;224:695–704. https://doi.org/10.1093/infdis/jiaa785.
Correa-Macedo, W, Fava, VM, Orlova, M, Cassart, P, Olivenstein, R, Sanz, J et al. Alveolar macrophages from persons living with HIV show impaired epigenetic response to Mycobacterium tuberculosis. J Clin Invest. 2021;131: https://doi.org/10.1172/jci148013.
McHenry, ML, Simmons, J, Hong, H, Malone, LL, Mayanja-Kizza, H, Bush, WS et al. Tuberculosis severity associates with variants and eQTLs related to vascular biology and infection-induced inflammation. medRxiv, 2022.2008.2023.22279140. 2022; https://doi.org/10.1101/2022.08.23.22279140.
Eckhardt M, Zhang W, Gross AM, Von Dollen J, Johnson JR, Franks-Skiba KE, et al. Multiple routes to oncogenesis are promoted by the human papillomavirus-host protein network. Cancer Discov. 2018;8:1474–89. https://doi.org/10.1158/2159-8290.cd-17-1018.
Zak DE, Aderem A. Systems biology of innate immunity. Immunol Rev. 2009;227:264–82.
Benedict KF, Lauffenburger DA. Insights into proteomic immune cell signaling and communication via data-driven modeling. Curr Top Microbiol Immunol. 2013;363:201–33. https://doi.org/10.1007/82_2012_249.
Ideker T, Krogan NJ. Differential network biology. Mol Syst Biol. 2012;8:565. https://doi.org/10.1038/msb.2011.99.
Mitra K, Carvunis AR, Ramesh SK, Ideker T. Integrative approaches for finding modular structure in biological networks. Nat Rev Genet. 2013;14:719–32. https://doi.org/10.1038/nrg3552.
Ashworth J, Wurtmann EJ, Baliga NS. Reverse engineering systems models of regulation: discovery, prediction and mechanisms. Curr Opin Biotechnol. 2012;23:598–603. https://doi.org/10.1016/j.copbio.2011.12.005.
Imam S, Schäuble S, Brooks AN, Baliga NS, Price ND. Data-driven integration of genome-scale regulatory and metabolic network models. Front Microbiol. 2015;6:409. https://doi.org/10.3389/fmicb.2015.00409.
Peterson EJ, Bailo R, Rothchild AC, Arrieta-Ortiz ML, Kaur A, Pan M, et al. Path-seq identifies an essential mycolate remodeling program for mycobacterial host adaptation. Mol Syst Biol. 2019;15:e8584. https://doi.org/10.15252/msb.20188584.
Haks MC, Bottazzi B, Cecchinato V, De Gregorio C, Del Giudice G, Kaufmann SHE, et al. Molecular signatures of immunity and immunogenicity in infection and vaccination. Front Immunol. 2017;8:1563. https://doi.org/10.3389/fimmu.2017.01563.
Fong LE, Muñoz-Rojas AR, Miller-Jensen K. Advancing systems immunology through data-driven statistical analysis. Curr Opin Biotechnol. 2018;52:109–15. https://doi.org/10.1016/j.copbio.2018.03.009.
Köhler S, Bauer S, Horn D, Robinson PN. Walking the interactome for prioritization of candidate disease genes. Am J Hum Genet. 2008;82:949–58. https://doi.org/10.1016/j.ajhg.2008.02.013.
Schurz, H, Naranbhai, V, Yates, TA, Gilchrist, JJ, Parks, T, Dodd, PJ et al. Multi-ancestry meta-analysis of host genetic susceptibility to tuberculosis identifies shared genetic architecture. medRxiv, 2022.2008.2026.22279009. 2022; https://doi.org/10.1101/2022.08.26.22279009.
Horne DJ, Graustein AD, Shah JA, Peterson G, Savlov M, Steele S, et al. Human ULK1 variation and susceptibility to mycobacterium tuberculosis infection. J Infect Dis. 2016;214:1260–7. https://doi.org/10.1093/infdis/jiw347.
Shah JA, Musvosvi M, Shey M, Horne DJ, Wells RD, Peterson GJ, et al. A functional toll-interacting protein variant is associated with bacillus calmette-guérin-specific immune responses and tuberculosis. Am J Respir Crit Care Med. 2017;196:502–11. https://doi.org/10.1164/rccm.201611-2346OC.
Hall NB, Igo RP Jr, Malone LL, Truitt B, Schnell A, Tao L, et al. Polymorphisms in TICAM2 and IL1B are associated with TB. Genes Immun. 2015;16:127–33. https://doi.org/10.1038/gene.2014.77.
Thye T, Browne EN, Chinbuah MA, Gyapong J, Osei I, Owusu-Dabo E, et al. IL10 haplotype associated with tuberculin skin test response but not with pulmonary TB. PLoS One. 2009;4:e5420. https://doi.org/10.1371/journal.pone.0005420.
Cobat A, Gallant CJ, Simkin L, Black GF, Stanley K, Hughes J, et al. Two loci control tuberculin skin test reactivity in an area hyperendemic for tuberculosis. J Exp Med. 2009;206:2583–91. https://doi.org/10.1084/jem.20090892.
Igo RP Jr., Hall NB, Malone LL, Hall JB, Truitt B, Qiu F, et al. Fine-mapping analysis of a chromosome 2 region linked to resistance to Mycobacterium tuberculosis infection in Uganda reveals potential regulatory variants. Genes Immun. 2018. https://doi.org/10.1038/s41435-018-0040-1.
Cobat A, Hoal EG, Gallant CJ, Simkin L, Black GF, Stanley K, et al. Identification of a major locus, TNF1, that controls BCG-triggered tumor necrosis factor production by leukocytes in an area hyperendemic for tuberculosis. Clin Infect Dis. 2013;57:963–70. https://doi.org/10.1093/cid/cit438.
Funding
Dr. Stein was supported by NIH grants 75N93019C00071 (to Fortune, Flynn, and Boom), RO1-AI147319 (to Bark, Whalen, and Kiwanuka), and U19AI162583 (to Hawn and Cox).
Author information
Authors and Affiliations
Contributions
All contributions were from the single author.
Corresponding author
Ethics declarations
Competing interests
The author declares no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Stein, C.M. Genetic epidemiology of resistance to M. tuberculosis Infection: importance of study design and recent findings. Genes Immun 24, 117–123 (2023). https://doi.org/10.1038/s41435-023-00204-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41435-023-00204-z
