
Abstract
Having pets in the house during the first years of life has been shown to protect against allergies. However, the result of different studies is heterogeneous. The aim of this study was to evaluate the methylation pattern in cord blood in relation to pet ownership during pregnancy.
We investigated the methylation patterns of 96 cord blood samples, participants of the Epigenetic Hallmark of Maternal Atopy and Diet—ELMA project, born to mothers who either owned pets (n = 32) or did not own pets (n = 64) during their pregnancy. DNA from cord blood was analysed using the Infinium methylation EPIC. For statistical analysis, RnBeads software was applied.
We found 113 differentially methylated sites (DMs) in the covariate-adjusted analysis (FDR p < 0.05), with small methylation differences. The top DMs were associated with genes: UBA7, THRAP3, GTDC1, PDE8A and SBK2. In the regional analysis, two promoter regions presented with significance: RN7SL621P and RNU6-211P. Cis-regulatory element analysis revealed significant associations with several immune-related pathways, such as regulation of IL18, Toll signalling, IL6 and complement.
We conclude that pet exposure during pregnancy causes subtle but significant changes in methylation patterns in cord blood, which are reflected in the biological processes governing both innate and adaptive immune responses.
Similar content being viewed by others


Background
Immunological status related to allergy could be determined as early as in utero. Environmental exposures at this particular moment in life are important for the development of a properly functioning immune system. Having pets during the first years of life has been shown to be protective against experiencing allergy at an age later in life. The effect seems to be dose-dependent, species non-specific and correlates with the number of animals present. This suggests that the effect results from extensive microbial and endotoxin exposure [1]. Having pets has been shown to be protective against asthma, allergic rhinitis, atopic dermatitis and sensitisation to aeroallergens, from both animals and pollens [2].
However, the results of studies are heterogeneous, with some showing an opposite effect. For example, household cat or dog exposure has been shown to associate with asthma in Chinese children [3]. Similarly, a meta-analysis of 11 birth cohorts did not reveal any significant association between having different kinds of pets and suffering from allergic diseases such as asthma, allergic rhinitis and atopic dermatitis, but showed protection against sensitisation to aeroallergens at age two in children exposed to furry animals at six months of age [4]. In some studies, the “pet effect” seemed to be species-specific. Cat ownership in the first year of life was revealed to be associated with a decrease in cat allergy and hay fever at age 13, while a dog in the household had no effect [5]. Different results for cats and dogs were also reported in adult populations, and the effect depended on coexistence with other risk factors [6]. A negative impact of pet keeping has been demonstrated in a few studies. An inverse correlation was described in a birth cohort where cat exposure corresponded with early onset of atopic dermatitis, with an interaction between filaggrin gene mutation (R501x) and cat exposure [7]. Epigenetic mechanism regarded as the main regulator of gene expression and the mirror of environmental exposure could be the key element in translating the effect of pet exposure early in life on the development of allergic disease.
The “pet effect” could be considered as a reflection of the effect observed in farming. Thus, the hypothesis could be proposed that household pets act as a “mini-farm”. For farming, there is strong evidence linking the methylation of different genes with protection against allergy. The top candidates are the CD14 promoter, ORMDL1, STAT6, RAD50 and IL13 [8]. There are few studies concerning prenatal exposure to pets and allergy outcomes in children. Until now, no effect was shown for either sensitisation or allergic rhinitis [9].
Previously, we have published an analysis regarding maternal atopy as the main exposure affecting offspring DNA methylation at birth [10]. Here we hypothesise that pet exposure in pregnancy influences DNA methylation in cord blood and this mechanism explains the existing pet effect on allergy occurrence.
Material and methods
Study group
We investigated methylation patterns in the cord blood of 96 newborns, which were children of women aged 25–34, living in an urban area, and participating in project Epigenetic Hallmark of Maternal Atopy and Diet (ELMA). Thirty-two of the women reported keeping animals at home while pregnant: 16 of them were keeping a cat, 14 had a dog, 10 had more than one animal, one had birds, and three had furred rodents. ELMA is an ongoing project designed to establish an epigenetic pattern related to maternal atopy and diet. The characteristics of the study group and analysis regarding maternal atopy were presented in our previous publication [10]. All women were recruited in the 3rd trimester, they were not smoking or exposed to ETS and did not report diabetes, hypertension or pre-pregnancy obesity. The study was approved by the Ethical Committee of the Wroclaw Medical University and all participants signed informed consent forms.
Methylome analysis
DNA from 96 cord blood samples was analysed using the Infinium MethylationEPIC Kit (Illumina, San Diego, CA) according to the standard protocol. All the procedures of sampling, DNA isolation, quality control are described in our previous study [10].
The applied Infinium MethylationEPIC array covers over 850,000 methylation sites scattered across the human genome. Apart from sites within known CpG islands, it includes probes identifying CpG sites outside CpG islands, non-CpG methylated sites identified in human stem cells (CHH sites), differentially methylated sites identified in tumour versus normal cells, FANTOM5 enhancers, ENCODE open chromatin and enhancers, DNase hypersensitive sites, and sites located in miRNA promoter regions.
Data analysis
Data analysis was performed RnBeads 2.4.0 software [11], as in our previous study [10] (and included probes filtering (based on missing data, detection p-value, location on sex chromosomes), normalisation (BMIQ method), bath effect removal (surrogate variable analysis) and covariates analysis. The only difference was the covariates used, which included the mother’s atopy, gestational age and sex. All covariates were included in a final analysis in which the refFreeEWAS method [12] was used for beta-value (methylation value) corrections regarding cell-type heterogeneity and the inclusion of covariates. The refFreeEWAS method allows reference-free deconvolution, providing proportions of putative cell types defined by their underlying methylomes and allowing explicit quantitation of the mediation of phenotypic associations with DNA methylation by cell composition effects. Finally, both FDR-corrected p-values from the refFreeEWAS model (statistical significance) and a RnBeads software score were used to independently detect two sets of sites that were differentially methylated (DM) between the groups considered. The RnBeads score which is originally designed and recommended by the software authors combines initial statistical testing (statistical significance) with a priority ranking based on the absolute and relative effect size of the differences between groups, assigning a combined rank score for differential DNA methylation to each analysed CpG site and genomic region [13].
The detailed analysis of gene content was performed for sites (CpGs) that were differentially methylated between groups with refFreeEWAS model FDR < 0.05. Additionally, to make use of the RnBeads rank (score), the top 100 (approximately top 0.01% of all sites) best-ranked sites were analysed, as well as the top 100 differentially methylated regions (DMRs). This cutoff was much more restrictive than the one proposed in the RnBeads software (the auto.select.rank.cut software function, which selected 126,646 sites and 1008 DMRs). DMRs were defined only within promoters as having the greatest potential to regulate genes expression through DNA methylation. Ensembl gene definitions were used for that, defining promoter regions as the 1500 bases upstream (TSS1500) and 500 bases downstream of the transcription start sites of corresponding genes. The genes associated with DM sites and DMRs were analysed using the WEB-based GEne SeT AnaLysis Toolkit [14] to identify enriched biological processes, pathways and human disease phenotypes. Overrepresentation tests were performed with respect to all known genes using an FDR correction for multiple testing [15]. Additional analysis was done using GREAT 4.0.4 software [16], which assigns biological meaning to a set of non-coding genomic regions by analysing the annotations of the nearby genes (with default settings for region association to genes). Thus, it is particularly useful for studying cis functions of sets of non-coding genomic regions (cis-regulatory elements). The resulting genes were tested for overrepresentation using standard settings of GREAT software, according to GO categories as well as human phenotype annotations.
Results
The comparison of the basic characteristics of the study groups is presented in Table 1.
Analysis of global methylation profile
As a final outcome of the filtering procedures, 22,423 probes and none of the samples were removed (96 samples and 827,101 probes were retained). Initial analysis detecting associations between principal components of the methylation profile and covariates showed that global methylation profile was only affected by gestational age and sex. Maternal atopy did not show a significant effect on global methylation profile, but it was associated with subtler methylation changes, as we previously described [10]. All confounding factors were used for β-value correction in the final statistical analysis. Additionally, a correction for cell-type heterogeneity was implemented as proposed by Houseman et al. (2014) [17]. The performed PCA showed a rather low level of global methylation profile variation among the study groups, with a portion of the samples from both groups having clearly distinct methylation profiles. Additional hierarchical clustering based on Euclidean distance confirmed the rather poor agreement of global methylation profile clustering and sample division into study groups, suggesting more discrete methylation changes caused by pet-keeping during pregnancy (Supplementary file 1).
Differential methylation analysis of single CpG sites
Differential methylation analysis of specific CpG sites revealed that, at the genome-wide level (after FDR correction for multiple testing), β-values of 113 sites differed significantly (FDR < 0.05) between the study groups (Table 2, Supplementary file 2). Analysis of the distribution of the sites across the genome showed that most (65%) were located in non-coding genome regions and outside annotated CpG islands (78%). The remaining sites were predominantly located in gene bodies (17%) or TSS1500 (11%). Of the sites located in the annotated CpG islands (n = 25), 13 (52%) were located in island cores, while the remaining sites were found in island shelves and shores. The average absolute delta-β for the differentially methylated (DM) sites was low with a value of 0.0044. Of the DM sites, 56 were hypomethylated in samples obtained from pet owners children and 57 were hypermethylated. The average delta-beta was slightly lower for hypomethylated sites (0.0037) than for hypermethylated sites (0.0052). The distribution of hyper- and hypomethylated DM sites in different functional elements did not differ significantly between groups (Supplementary file 3).
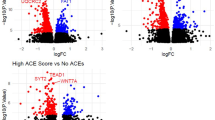
Additional analysis was dedicated to the most differentially methylated sites according to RnBeads software rank (score) (Table 3, Supplementary file 4). This was due to the fact that none of the top-ranked sites were identified as significant after FDR correction. Analysis of the distribution of top-ranked sites across the genome revealed that most of them were located in non-coding genome regions (63%) and outside CpG islands (72%). Of all sites, 21% were located in gene bodies, 14% in gene promotes and 13% within islands cores. The average absolute difference in methylation between study groups for ranked sites was 0.0427, tenfold higher than for FDR-detected sites. Most of the sites (60%) were hypomethylated in the group of pet owners’ children (delta-beta = 0.042). The hypermethylated sites had a similar difference in methylation to the hypomethylated sites. Statistical analysis of the distribution of hyper- and hypomethylated CpG sites showed that they were differentially distributed (p = 0.009) within CpG islands. Among the hypomethylated sites, there were more sites located outside known islands (83% vs. 55%), while hypermethylation was more common in the islands cores (27% vs. 3%) (Supplementary file 3). The mutual relationship between FDR-detected and rank-detected DM sites can be seen in Fig. 1.

Color scale according to combined ranking.The mutual relationship between FDR-detected and rank-detected DM sites.
Differentially methylated regions
Regional analysis was restricted to gene promoters in this study. An FDR-based threshold detected only two significant (FDR < 0.05) genome regions, located on chromosomes 5 and 8 (Supplementary file 5). One of the regions was hypo- and another was hypermethylated. The regions were associated with the putative promoters of RN7SL621P (hypomethylated in pet owners) and RNU6-211P (hypermethylated). The rank-based analysis allowed us to analyse the DMRs, of which 47% were hypermethylated in pet owners’ children. The average methylation difference for those sites was 0.018. The delta-beta for hypomethylated sites was similar (0.016) (Supplementary file 6).
Analysis of genes associated with DM sites and DMRs
The 113 DM sites detected by FDR were annotated to 82 different genes (Supplementary file 2). The genes did not enrich any biological processes after FDR but showed pointwise enrichments for processes associated with positive regulation of B cell-mediated immunity and positive regulation of immunoglobulin-mediated immune response (C17orf99, PTPRC, TP53BP1). Pointwise enrichments were also detected for some KEGG pathways, such as ascorbate and aldarate metabolism, cortisol synthesis and secretion, and Fc gamma R-mediated phagocytosis. Among detected disease phenotypes, there were none with significance after FDR and they were difficult to associate with the expected phenotype (Supplementary file 2). Additional GREAT analysis of cis-regulatory elements did not reveal any significantly enriched GO biological processes or phenotypes, however, it showed pointwise significance for epithelial structure maintenance (MKS1, CROCC, SOX9, MUC4) and negative regulation of ERAD pathway (USP25, UBXN1), amongst others (Supplementary file 2).
Sites detected as DM by RnBeads rank were annotated to 59 different genes (Supplementary file 4). The analysis of their associated biological processes did not show significant enrichment of any categories, however, some enrichment was found in processes such as positive regulation of amyloid precursor protein catabolic process, regulation of amyloid-beta formation (GSAP, SPON1), membrane lipid catabolic process (ENPP7, PNLIPRP2) and regulation of angiogenesis (ADAM12, IL17F, PRKCB, PTPRM) (Supplementary file 4). The associated KEGG pathways also did not show statistical significance after correction for multiple testing, however, point enrichments were found for insulin secretion, aldosterone synthesis and secretion, steroid hormone biosynthesis (CACNA1C, CREB5, PRKCB), cortisol synthesis and secretion (CACNA1C, CREB5), and others (Supplementary file 4). However, the GREAT analysis revealed FDR-corrected significant biological processes such as negative regulation of complement activation, lectin pathway (A2M) (FDR = 1.10 × 10−8), negative regulation of immune response (A2M, IL1RL1, TRIM27, FOXF1, HLA-G, DUSP22) (FDR = 4.92 × 10−5), cellular defense response (KLRG1, IL1RL2, HLA-G) (FDR = 5.32 × 10−5), and primary lung bud formation (RDH10) (FDR = 4.67 × 10−5), amongst others (Supplementary file 4).
Region-based analysis using FDR showed only two associated genes, RN7SL621P, and RNU6-211P. Thus, enrichment analysis was applied to the top DM regions classified by RnBeads rank. These regions were associated with 62 different genes. No significant enrichment of biological processes was found for the genes while using FDR, however, uncorrected p-values were significant for processes associated with regulation of leucocyte tethering or rolling (CCL25, CCR2), regulation of cytokine production (CCR2, HLA-DPA1, MAPK13, MAST2, SMAD3, SULF1) and regulation of leucocyte adhesion to vascular endothelial cell (CCL25, CCR2) (Supplementary file 6). Point significance was also found for KEEG pathways such as Th17 cell differentiation (HLA-DPA1, MAPK13, SMAD3), the intestinal immune network for IgA production (CCL25, HLA-DPA1), inflammatory bowel disease (HLA-DPA1, SMAD3), and Th1/Th2 cell differentiation (HLA-DPA1, MAPK13). Cis-regulatory element analysis by GREAT software showed some other immune-related biological processes associated with the analysed regions, including regulation of interleukin-18 biosynthetic process (FDR = 1.18 × 10−54), negative regulation of Toll signalling pathway (NLRP12) (FDR = 1.18 × 10−51), negative regulation of interleukin-6 biosynthetic process (NLRP12, GHRL, FOXJ1, GHSR, INPP5D) (2.88 × 10−24), and antigen processing and presentation of exogenous peptide antigen via MHC class I, TAP-independent (B2M, HLA-B, HLA-F, HLA-G, LNPEP) (FDR = 9.77 × 10−16), amongst others. The genes associated with DMRs (by GREAT analysis) also showed enrichment of human phenotypes connected with elevated C-reactive protein level (NLRP12) (FDR = 2.40 × 10−51), recurrent aphthous stomatitis (IL17RC, ITK, NCF4, NLRP12, NLRP3, ORAI1, PRKDC) (FDR = 1.19 × 10−29), progressive gait ataxia (C10orf2, POLG, SACS, SETX, UBE3A) (FDR = 8.00 × 10−26), and sleep–wake cycle disturbance (APOE, DHCR7, PER2, PSEN2, UBE3A) (FDR = 5.33 × 10−25) (Supplementary file 6).
Discussion
Environmental exposures during pregnancy, among them pet exposure are believed to have an impact on offspring DNA methylation patterns, further affecting the development of a child’s immune system. This is the perspective relates to foetal programming of allergic diseases. There is evidence that animal exposure could protect against allergy, specifically if it occurs early in life. The mechanism of such protection is poorly understood but epigenetics could be the key in that process. We have studied global DNA methylation patterns in cord blood of neonates born to mothers who either owned or did not pets during pregnancy and recognised multiple DMs in relation to this exposure. According to the standard procedure, we have identified 113 DMs and two DMRs with FDR-adjusted p-values < 0.05. Overall, the methylation differences were small. The top DMs corresponded to genes that were revealed to be associated with asthma in other EWAS study, such as GTDC1, SNCAIP, CDC42BPB, AMPH, AMBRA1, MIR99AHG, SLC7A11—revealed as DMs in lung tissues. The probes which were assigned as DMs in our analysis were with a different location than those described in other studies, though annotated to the same genes [18, 19]. Among top DMs, there was also UBA7 which is related to the pathways of the innate immune system [20], and THRAP3 is linked to circadian rhythm genes [21]—component of this molecular clock has been shown to be disrupted in asthma. Other gene from the top 30 —PDE8 is expressed in the smooth muscle of the airway and regulates the response to beta-adrenergic receptors, so it has been proposed as a novel therapeutic target for asthma remodelling [22]. VEGFA gene has been revealed as associated with postnatal lung development. VEGF itself has been implicated in remodelling and asthma development [23]. In the regional analysis, we identified two promoter regions with significant methylation differences (FDR-adjusted). Both are related to pseudogenes with unknown functions. In pathway analysis, the genes corresponding to the DMs and DMRs did not enrich significantly any biological processes. However, a trend was observed for regulation of B cell immunity, immunoglobulin production and epithelial structure (in the analysis of individual sites), and cytokine production, Th17 differentiation, B cell immunity and Th1/Th2 differentiation (in regional analysis).
All of these processes may be related to the early events in immune switching occurring as a consequence of pet exposure. Specifically, Th17 may play a role. Th17 cells have the same precursor as Tregs, which are known for their induction of immune tolerance. While Tregs inhibit inflammation, Th17 cells activate processes involved in the immune response against bacteria and fungi. Segmented filamentous bacteria induce Th17, while Bacteroides and Clostridia induce Tregs [24]. The Th17/Treg balance may be important in the context of pet exposure, which is proposed to be a so-called “mini-farm” environment, with a corresponding diversity of microbial stimuli. The same process has been observed in animal models where specific microbial exposure has been shown to be connected to prenatal priming of the acquired asthmatic resistant phenotype [25]. We observe only a trend here, which requires further evaluation in the future. Also, cis-regulatory element analysis did not reveal any significant association for the DMRs and DMs identified with an FDR-corrected p-value cutoff.
According to the RnBeads scores, the top-ranking sites were CpG annotated to the genes: RNTF2, LOC102467224, ICMT, ANK1, and TRABD2B. RNTF2 is of special interest. This gene has been shown to be associated with innate immune processes and IL-3 responses related to inflammation. Priming of RNTF2 with LPS (lipopolisacharyde), in mice exposed to bacteria (Pseudomonas), results in receptor abundance, causing lung inflammation. Conversely, overexpression of RNTF2 is linked to reductions in these processes [26]. ICMT and ANK1 are proteins with functional connections to cell membranes, making them potential candidates important for the integrity of epithelial barriers. The disruption of this integrity is believed to be the first step in allergen sensitisation. TRABD2B is a gene encoding a metalloproteinase involved in Wnt signalling, which regulates processes including fibrosis and smooth muscle hypertrophy—phenomena observed in asthma [27]. The 100 top-ranking (by RnBead score) DMs did not enrich significantly any biological process. However, some were identified in another EWAS study regarding respiratory allergy and the methylation profile of saliva: cg13575925, cg04155231, cg23795048 and cg14089881(p < 0.05, |Δβ| > 0.2) [28]. The first three map to the gene LOC14457, encoding a long non-coding RNA, and the last maps to ICMT [28].
Cis-regulatory element analysis of the 100 top-ranking DMs (by RnBead score), showed significant enrichment for important biological processes potentially associated with allergy, including negative regulation of complement activation connected to A2M gene, negative regulation of immune response related to genes (A2M, IL1RL1, TRIM27, FOXF1, HLA-G, DUSP22), negative regulation of cellular defence (KLRG1, IL1RL2, HLA-G), and lung formation (RDH10). KLRG1 was the most significant gene in this analysis.
The complement system constitutes a link between innate and adaptive immune responses. Complement components C3a and C5a have been shown to take part in type I hypersensitivity responses [29]. 4,5-fold increase in expression of the A2M protein has been revealed by proteome analysis in the nasal mucosa of patients with allergic rhinitis [30]. Additionally, elevated serum A2M levels have been seen in allergic rhinitis patients, correlating with IL17 level and nasal congestion [31]. For IL1RL1, there are some studies concerning genetic variants and methylation in regard to asthma, however, with rather opposite results [32, 33]. Also, some polymorphisms in this gene seem to be associated with specific types of food allergy in children (peanut and egg/chicken) [34]. Nevertheless, IL33/IL1RL1 is thought to be a possible therapeutic target for asthma treatment [35]. Another gene product, TRIM27, takes part in the negative regulation of mast cells, crucial for the allergic response [36]. HLA-G could also suppress allergic inflammation [37], but soluble particles of HLA-G act as a modulator of allergic responses [38] KLRG1 is a marker of ILC cells, which are important components of the innate immune response. ILC cells presenting KLRG1 predominantly differentiate into ILC2-type cells, which exhibit functional characteristics of the type 2 response (independently of Th2), and are also a cellular source of IL5 and IL13. The role of these cells has been demonstrated in the HDM model of chronic asthma and IL9-derived, allergen-induced inflammation [39].
In the regional analysis, LINC00612, A2M-AS1, SULF1, MIR4766 and RNA55P253 were the top-ranking with regard to the RnBeads score. LINC00612 is a non-coding RNA, taking part in apoptosis, inflammation and oxidative stress, and also in the pulmonary microvascular response to ETS [40]. A2M-AS1 is a long non-coding RNA with a known function in chronic obstructive pulmonary disease (COPD) [41]. SULF1 is associated with lung development and endothelial function linked to inflammatory processes in the lungs [42]. The genes corresponding to the 100 top-ranking DMRs by RnBeads score did not enrich significantly any biological processes; the only trend observed was for the intestinal network for IgA production, and Th17, Th1 and Th2 cell differentiation—processes with possible links to food allergy.
In cis-regulatory element analysis for top-ranking DMRs by RnBeads score, we have found significant enrichment in several biological processes which possibly link the environmental exposure to the switch in immune status. Among these processes were: negative regulation of IL18, negative regulation of Toll signalling (NLRP12), and negative regulation of IL6 (NLRP12, GHRL, FOXJ1, GHBR, INPP4D). IL18 belongs to the IL1 family and its primary function is linked to an increase in the production of IFN gamma and Th1 maturation [43]. However, in some other studies, IL18 seems to cooperate with IL5 to produce eosinophilic inflammation in asthma [44]. The exact function of this cytokine may depend on the presence of other cytokines.
Negative regulation of Toll signalling is of special interest regarding exposure to the “pet environment”. This process seems to be directly connected to microbial stimuli. NLRP12 belongs to the family of NOD-like receptors, functioning as an attenuating factor of inflammation by suppressing inflammatory responses in activated monocytes. The exact role of NOD receptors is not well understood. NOD-1 and NOD2 activate Th1 and Th17 cells with increased production of TNF and IL1. SNPs within the corresponding genes have been shown to be associated with childhood asthma, while SNPs within NLRP12 are related to atopic dermatitis. Toll-like receptors play a dual role. They suppress allergy on activation by microbes, but they can also activate the response to allergens [45, 46]. Their presence and activity may be modulated by “animal” allergens [47]. Both NOD signalling and Toll signalling are part of the innate immune response to microbes. This constitutes the link between pet exposure (and the associated increased exposure to microbes) and the change in immune status.
Another component with regard to cis-regulatory elements analysis—IL6 is a typical inflammatory cytokine, taking part in the immune response to viruses, with enhanced production seen in COVID-19 disease Its role in asthma is not well established, but elevated levels have been seen in severe cases. ORMDL3, which has a known role in asthma pathogenesis, activates the IL6 pathway [48]. Among the genes affecting IL6 is FOXJ1, which has a role related to cilia function in the respiratory tract, and is also associated with primary ciliary dyskinesia (PCD) [49].
The evidence for the possible impact of pet-keeping during pregnancy on the methylation pattern in the newborn is scarce, with only a few studies addressing this problem. In one recent study, constant pet ownership from birth to secondary school was associated with changes in methylation status at cg03565274 and was shown to be protective against the combined phenotype asthma and/or rhinitis. According to eQTM analysis, methylation at this site corresponded to ZYMND10 gene expression. This gene is primarily responsible for PCD, a disease related to the abnormal structure of the cilia and recurrent respiratory tract infections. The connection with asthma or rhinitis from a pathomechanistic point of view is not clear [50]. Similarly, methylation status at CD14 has been reported to be associated with pet-keeping throughout childhood. CD14 is known for its role in the response to the microbiome, with altered expression in children raised on a farm [51].
Atopic parents tend to avoid having pets at home. Thus, reverse causality is sometimes considered as an explanation for why pet-keeping seems to protect against allergy. We addressed this problem by adjusting for maternal atopy in the current analysis.
Our study has some limitations. The sample size is small and we did not replicate the study in another cohort. However, the design of this study was based on a unique group with strictly defined risk factors, thus making finding an available cohort for replication difficult. There is not any similar study performed in 850K platform.
Conclusion
We concluded that pet exposure during pregnancy causes subtle but significant changes in methylation patterns, which are reflected in changes in the biological processes governing both the innate (Toll signalling, ILC, complement) and adaptive (IL18, IL6) immune responses. The functional relationship of this alternation requires further research, however, the switch in immunological status appears to be a plausible link between the pet exposure in pregnancy and allergy outcome in offspring
References
Hesselmar B, Hicke-Roberts A, Lundell AC, Adlerberth I, Rudin A, Saalman R, et al. Pet-keeping in early life reduces the risk of allergy in a dose-dependent fashion. PLoS ONE. 2018;13:1–14.
Ojwang V, Nwaru BI, Takkinen HM, Kaila M, Niemelä O, Haapala AM, et al. Early exposure to cats, dogs and farm animals and the risk of childhood asthma and allergy. PubMed - NCBI [Internet]. Pediatric Allergy Immunol. 2019;31:265–72. http://www.ncbi.nlm.nih.gov/pubmed/31829464.
Song N, Mohammed S, Zhang J, Wu J, Fu C, Hao S, et al. Prevalence, severity and risk factors of asthma, rhinitis and eczema in a large group of Chinese schoolchildren. J Asthma. 2014;51:232–42.
Lødrup Carlsen KC, Roll S, Carlsen KH, Mowinckel P, Wijga AH, Brunekreef B, et al. Does Pet Ownership in Infancy Lead to Asthma or Allergy at School Age? Pooled Analysis of Individual Participant Data from 11 European Birth Cohorts. PLoS ONE. 2012;7:e43214.
Al-Tamprouri C, Malin B, Bill H, Lennart B, Anna S. Cat and dog ownership during/after the first year of life and risk for sensitization and reported allergy symptoms at age 13. Immun Inflamm Dis. 2019;7:250–7. http://www.ncbi.nlm.nih.gov/pubmed/31464382.
Svanes C, Heinrich J, Jarvis D, Chinn S, Omenaas E, Gulsvik A, et al. Pet-keeping in childhood and adult asthma and hay fever: European community respiratory health survey. J Allergy Clin Immunol. 2003;112:289–300. http://www.ncbi.nlm.nih.gov/pubmed/12897734.
Thyssen JP, Ahluwalia TS, Paternoster L, Ballardini N, Bergström A, Melén E, et al. Interaction between filaggrin mutations and neonatal cat exposure in atopic dermatitis. Allergy. 2020;5:1481–5.
Michel S, Busato F, Genuneit J, Pekkanen J, Dalphin JC, Riedler J, et al. Farm exposure and time trends in early childhood may influence DNA methylation in genes related to asthma and allergy. Allergy Eur. J Allergy Clin Immunol. 2013;68:355–64.
Schoos AMM, Chawes BL, Jelding-Dannemand E, Elfman LB, Bisgaard H. Early indoor aeroallergen exposure is not associated with development of sensitization or allergic rhinitis in high-risk children. Allergy Eur. J Allergy Clin Immunol. 2016;71:684–91. http://www.ncbi.nlm.nih.gov/pubmed/26836471.
Danielewicz H, Gurgul A, Dębińska A, Myszczyszyn G, Szmatoła T, Myszkal A, et al. Maternal atopy and offspring epigenome-wide methylation signature. Epigenetics. 2020;1–13. https://www.tandfonline.com/doi/full/10.1080/15592294.2020.1814504?af=R&utm_source=researcher_app&utm_medium=referral&utm_campaign=RESR_MRKT_Researcher_inbound.
Müller F, Scherer M, Assenov Y, Lutsik P, Walter J, Lengauer T, et al. RnBeads 2.0: comprehensive analysis of DNA methylation data. Genome Biol. 2019;20:55. https://genomebiology.biomedcentral.com/articles/10.1186/s13059-019-1664-9.
Houseman EA, Kile ML, Christiani DC, Ince TA, Kelsey KT, Marsit CJ. Reference-free deconvolution of DNA methylation data and mediation by cell composition effects. BMC Bioinformatics 2016;17. http://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-016-1140-4.
Assenov Y, Müller F, Lutsik P, Walter J, Lengauer T, Bock C. Comprehensive analysis of DNA methylation data with RnBeads. Nat Methods. 2014;11:1138–40. http://www.nature.com/doifinder/10.1038/nmeth.3115%5Cnpapers3://publication//10.1038/nmeth.3115.
Wang J, Vasaikar S, Shi Z, Greer M, Zhang B. WebGestalt 2017: a more comprehensive, powerful, flexible and interactive gene set enrichment analysis toolkit. Nucleic Acids Res. 2017;45:W130–7.
Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B 1995;57:289–300.
McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, et al. GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol. 2010;28:495–501. https://www.nature.com/articles/nbt.1630.
Houseman EA, Molitor J, Marsit CJ. Reference-free cell mixture adjustments in analysis of DNA methylation data. Bioinformatics. 2014;30:1431–9. https://academic.oup.com/bioinformatics/article-lookup/.
Nicodemus-Johnson J, Myers RA, Sakabe NJ, Sobreira DR, Hogarth DK, Naureckas ET, et al. DNA methylation in lung cells is associated with asthma endotypes and genetic risk. JCI Insight. 2016;1. PMC5139904.
Li M, Zou D, Li Z, Gao R, Sang J, Zhang Y, et al. EWAS Atlas: a curated knowledgebase of epigenome-wide association studies. Nucleic Acids Res. 2019;47:D983–8. http://www.ncbi.nlm.nih.gov/pubmed/30364969.
Çalışkan M, Baker SW, Gilad Y, Ober C. Host genetic variation influences gene expression response to rhinovirus infection. PLoS Genet. 2015;11:1–15.
Marcheva B, Perelis M, Weidemann BJ, Taguchi A, Lin H, Omura C, et al. A role for alternative splicing in circadian control of exocytosis and glucose homeostasis. Genes Dev. 2020;34:1089–105. https://pubmed.ncbi.nlm.nih.gov/32616519/.
Johnstone TB, Smith KH, Koziol-White CJ, Li F, Kazarian AG, Corpuz ML, et al. PDE8 is expressed in human airway smooth muscle and selectively regulates cAMP signaling by b2-adrenergic receptors and adenylyl cyclase 6. Am J Respir Cell Mol Biol. 2018;58:530–41. https://pubmed.ncbi.nlm.nih.gov/29262264/.
Kreiner-Møller E, Chawes BLK, Vissing NH, Koppelman GH, Postma DS, Madsen JS, et al. VEGFA variants are associated with pre-school lung function, but not neonatal lung function. Clin Exp Allergy. 2013;43:1236–45. https://pubmed.ncbi.nlm.nih.gov/24152156/.
Lee GR. The balance of th17 versus treg cells in autoimmunity. Int J Mol Sci. 2018;19:730.
Holt PG, Strickland DH, Custovic A. Targeting maternal immune function during pregnancy for asthma prevention in offspring: harnessing the “farm effect”? J Allergy Clin Immunol. 2020;146:270–2.
Tong Y, Lear TB, Evankovich J, Chen Y, Londino JD, Myerburg MM, et al. The RNFT2/IL-3Rα axis regulates IL-3 signaling and innate immunity. JCI Insight. 2020;5:e133652.
Kwak HJ, Park DW, Seo JY, Moon JY, Kim TH, Sohn JW, et al. The Wnt/β-catenin signaling pathway regulates the development of airway remodeling in patients with asthma. Exp Mol Med. 2015;47:e198. PMC4686695.
Langie SAS, Szic KS, Declerck K, Traen S, Koppen G, Van Camp G, et al. Whole-Genome saliva and blood DNA methylation profiling in individuals with a respiratory allergy. PLoS ONE. 2016;11:e0151109.
Gerard NP, Gerard C. Complement in allergy and asthma. Curr Opin Immunol. 2002;14:705–8. https://pubmed-1ncbi-1nlm-1nih-1gov-1yxc2mlit0336.han.bg.umed.wroc.pl/12413519/.
Tomazic PV, Birner-Gruenberger R, Leitner A, Obrist B, Spoerk S, Lang-Loidolt D. Nasal mucus proteomic changes reflect altered immune responses and epithelial permeability in patients with allergic rhinitis. J Allergy Clin Immunol. 2014;133:741–50. https://pubmed-1ncbi-1nlm-1nih-1gov-1yxc2mlit0336.han.bg.umed.wroc.pl/24290289/.
Chen X, Xie ZH, Lv YX, Tang QP, Zhang H, Zhang JY, et al. A proteomics analysis reveals that A2M might be regulated by STAT3 in persistent allergic rhinitis. Clin Exp Allergy. 2016;46:813–24. https://pubmed-1ncbi-1nlm-1nih-1gov-1yxc2mlit0336.han.bg.umed.wroc.pl/27228572/.
Dijk FN, Xu C, Melén E, Carsin AE, Kumar A, Nolte IM, et al. Genetic regulation of IL1RL1 methylation and IL1RL1—a protein levels in asthma. Eur Respir J. 2018;51. https://pubmed-1ncbi-1nlm-1nih-1gov-1yxc2mlit0336.han.bg.umed.wroc.pl/29519908/.
Queiroz GA, Costa RS, Alcantara-Neves NM, Nunes de Oliveira Costa G, Barreto ML, Carneiro VL, et al. IL33 and IL1RL1 variants are associated with asthma and atopy in a Brazilian population. Int J Immunogenet. 2017;44:51–61. https://pubmed-1ncbi-1nlm-1nih-1gov-1yxc2mlit0336.han.bg.umed.wroc.pl/28266165/.
Ketelaar ME, Westerlaken – van Ginkel CD, Nawijn MC, EJ Dubois A, Koppelman GH. IL‐1RL1a serum levels and IL1RL1 SNPs in the prediction of food allergy. Clin Exp Allergy. 2021;cea.13802. https://onlinelibrary.wiley.com/https://doi.org/10.1111/cea.13802.
Takatori H, Makita S, Ito T, Matsuki A, Nakajima H. Regulatory mechanisms of IL-33-ST2-mediated allergic inflammation. Front Immunol. 2018;9. https://pubmed.ncbi.nlm.nih.gov/30233590/.
Srivastava S, Cai X, Li Z, Sun Y, Skolnik EY. Phosphatidylinositol-3-kinase C2 and TRIM27 function to positively and negatively regulate IgE receptor activation of mast cells. Mol Cell Biol. 2012;32:3132–9. https://pubmed-1ncbi-1nlm-1nih-1gov-1yxc2mlit0336.han.bg.umed.wroc.pl/22645315/.
Murdaca G, Contini P, Negrini S, Ciprandi G, Puppo F. Immunoregulatory role of HLA-G in allergic diseases. J Immunol Res. 2016;2016. https://pubmed-1ncbi-1nlm-1nih-1gov-1yxc2mlit0336.han.bg.umed.wroc.pl/27413762.
Ciprandi G, DeAmici M. Soluble HLA-G serum levels depend on allergy type and IgE levels. Allergy Rhinol. 2014;5:ar.2014.5.0076. https://pubmed-1ncbi-1nlm-1nih-1gov-1yxc2mlit0336.han.bg.umed.wroc.pl/24612937.
Nagasawa M, Heesters BA, Kradolfer CMA, Krabbendam L, Martinez-Gonzalez I, De Bruijn MJW, et al. KLRG1 and NKp46 discriminate subpopulations of human CD117 + CRTH2- ILCs biased toward ILC2 or ILC3. J Exp Med. 2019;216:1762–76. PMC6683990.
Luo J, Li L, Hu D, Zhang X. Linc00612/mir-31-5p/notch1 axis regulates apoptosis, inflammation, and oxidative stress in human pulmonary microvascular endothelial cells induced by cigarette smoke extract. Int J COPD. 2020;15:2049–60.
Qian Y, Mao Z, Shi Y, Liu Z, Cao Q, Zhang Q. Comprehensive analysis of miRNA–mRNA–lncRNA networks in non-smoking and smoking patients with chronic obstructive pulmonary disease. Cell Physiol Biochem. 2018;50:1140–53. http://www.ncbi.nlm.nih.gov/pubmed/30355907.
Li X, Hawkins GA, Ampleford EJ, Moore WC, Li H, Hastie AT, et al. Genome-wide association study identifies TH1 pathway genes associated with lung function in asthmatic patients. J Allergy Clin Immunol. 2013;132:313–20.e15.
Nakanishi K. Unique action of Interleukin-18 on T cells and other immune cells. Front Immunol. 2018;9. https://pubmed-1ncbi-1nlm-1nih-1gov-1yxc2mlit0336.han.bg.umed.wroc.pl/29731751.
Xu MH, Yuan FL, Wang SJ, Xu HY, Li CW, Tong X. Association of interleukin-18 and asthma. Inflammation. 2017;40:324–7. https://pubmed-1ncbi-1nlm-1nih-1gov-1yxc2mlit0336.han.bg.umed.wroc.pl/27913952.
Zakeri A, Russo M. Dual role of toll-like receptors in human and experimental asthma models. Front Immunol. 2018;9. PMC5963123.
Monie TP, Bryant CE. Allergens and activation of the toll-like receptor response. Methods Mol Biol. 2016;341–50. https://pubmed-1ncbi-1nlm-1nih-1gov-1yxc2mlit039d.han.bg.umed.wroc.pl/26803639/.
Herre J, Grönlund H, Brooks H, Hopkins L, Waggoner L, Murton B, et al. Allergens as immunomodulatory proteins: the cat dander protein Fel d 1 enhances TLR activation by lipid ligands. J Immunol. 2013;191:1529–35.
Das S, Miller M, Broide DH. Chromosome 17q21 genes ORMDL3 and GSDMB in asthma and immune diseases. Adv Immunol. 2017;1–52. https://pubmed-1ncbi-1nlm-1nih-1gov-1yxc2mlit0458.han.bg.umed.wroc.pl/28826527/.
Mukherjee I, Roy S, Chakrabarti S, Mukherjee I, Roy S, Chakrabarti S. Identification of important effector proteins in the FOXJ1 transcriptional network associated with ciliogenesis and ciliary function. Front Genet. 2019;10. https://pubmed-1ncbi-1nlm-1nih-1gov-1yxc2mlit0458.han.bg.umed.wroc.pl/30881373/.
Qi C, Jiang Y, Yang IV, Forno E, Wang T, Vonk JM, et al. Nasal DNA methylation profiling of asthma and rhinitis. J Allergy Clin Immunol. 2020;145:1655–63.
Munthe-Kaas MC, Bertelsen RJ, Torjussen TM, Hjorthaug HS, Undlien DE, Lyle R, et al. Pet keeping and tobacco exposure influence CD14 methylation in childhood. Pediatr Allergy Immunol. 2012;23:746–53. http://www.ncbi.nlm.nih.gov/pubmed/23194293.
Funding
This study was funded by the National Science Centre, Poland 2015/19/B/NZ5/00041.
Author information
Authors and Affiliations
Contributions
HD, AB have contributed substantially to the conception and design of the work. HD, GM, AD, AM, AD-C have contributed to the acquisition of data. HD, AG, TSZ, IJ have contributed to the analysis of data. HD, AG, AD, GM, TSZ, AM, IJ, LH, AB have contributed to the interpretation of the data. HD, AG have contributed to the drafting of the manuscript. HD, AG, AD, GM, TSZ, AM, IJ, AD-C, LH, AB have contributed to revising the work.
Corresponding author
Ethics declarations
Competing interests
HD, AG, AD, GM, TS, AM, GJ, Drabik-Chamerska A report grant and personal fees from the National Science Center, Poland, during the conduct of the study; HD reports lecturer fees from Mead Johnson Nutrition, Takeda outside the submitted work; LH, AB report a grant from the National Science Center, Poland, during the conduct of the study.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Danielewicz, H., Gurgul, A., Dębińska, A. et al. Pet ownership in pregnancy and methylation pattern in cord blood. Genes Immun 22, 305–312 (2021). https://doi.org/10.1038/s41435-021-00151-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41435-021-00151-7

{kind=link}