
Abstract
Recombinant adeno-associated virus (rAAV) has become one of the most promising gene delivery systems for both in vitro and in vivo applications. However, a key challenge is the lack of suitable imaging technologies to evaluate delivery, biodistribution and tropism of rAAVs and efficiently monitor disease amelioration promoted by AAV-based therapies at a whole-organ level with single-cell resolution. Therefore, we aimed to establish a new pipeline for the biodistribution analysis of natural and new variants of AAVs at a whole-brain level by tissue clearing and light-sheet fluorescence microscopy (LSFM). To test this platform, neonatal C57BL/6 mice were intravenously injected with rAAV9 encoding EGFP and, after sacrifice, brains were processed by standard immunohistochemistry and a recently released aqueous-based clearing procedure. This clearing technique required no dedicated equipment and rendered highly cleared brains, while simultaneously preserving endogenous fluorescence. Moreover, three-dimensional imaging by LSFM allowed the quantitative analysis of EGFP at a whole-brain level, as well as the reconstruction of Purkinje cells for the retrieval of valuable morphological information inaccessible by standard immunohistochemistry. In conclusion, the pipeline herein described takes the AAVs to a new level when coupled to LSFM, proving its worth as a bioimaging tool in tropism and gene therapy studies.

This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout







Similar content being viewed by others


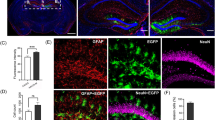
Data availability
All data generated or analysed during this study are included in this published article (and its supporting information files) or are available from the corresponding author on reasonable request.
References
Miyake K, Miyake N, Yamazaki Y, Shimada T, Hirai Y. Serotype-independent method of recombinant adeno-associated virus (AAV) vector production and purification. J Nippon Med Sch. 2012;79:394–402. https://www.jstage.jst.go.jp/article/jnms/79/6/79_394/_article.
Ojala DS, Amara DP, Schaffer DV. Adeno-associated virus vectors and neurological gene therapy. Neuroscientist. 2014;21:84–98. http://nro.sagepub.com/content/early/2014/02/19/1073858414521870.abstract.
Russell DW, Alexander IE, Miller AD. DNA synthesis and topoisomerase inhibitors increase transduction by adeno-associated virus vectors. Proc Natl Acad Sci USA. 1995;92:5719–23.
Atchison RW, Casto BC, Hammon WM. Adenovirus-associated defective virus particles. Science. 1965;149:754–6.
Muzyczka N. Use of adeno-associated virus as a general transduction vector for mammalian cells. In: Muzyczka N, editor. Viral Expression Vectors. Berlin, Heidelberg: Springer Berlin Heidelberg; 1992. 97–129. https://doi.org/10.1007/978-3-642-75608-5_5.
Grieger JC, Choi VW, Samulski RJ. Production and characterization of adeno-associated viral vectors. Nat Protoc. 2006;1:1412–28. http://www.nature.com/doifinder/10.1038/nprot.2006.207.
Gao G, Vandenberghe LH, Alvira MR, Lu Y, Calcedo R, Zhou X, et al. Clades of Adeno-associated viruses are widely disseminated in human tissues. J Virol. 2004;78:6381–8.
Gao G, Vandenberghe LH, Wilson JM. New recombinant serotypes of AAV vectors. Curr Gene Ther. 2005;5:285–97.
Van Vliet KM, Blouin V, Brument N, et al. The role of the adeno-associated virus capsid in gene transfer. In: Methods in molecular biology (Clifton, NJ). United States; 2008. 51–91. http://link.springer.com/10.1007/978-1-59745-210-6_2.
Murlidharan G, Samulski RJ, Asokan A. Biology of adeno-associated viral vectors in the central nervous system. Front Mol Neurosci. 2014;7:1–9. http://journal.frontiersin.org/article/10.3389/fnmol.2014.00076/abstract.
Foust KD, Nurre E, Montgomery CL, Hernandez A, Chan CM, Kaspar BK. Intravascular AAV9 preferentially targets neonatal neurons and adult astrocytes. Nat Biotechnol. 2009;27:59–65.
Hermonat PL, Muzyczka N. Use of adeno-associated virus as a mammalian DNA cloning vector: transduction of neomycin resistance into mammalian tissue culture cells. Proc Natl Acad Sci U S A. 1984;81:6466–70.
Tratschin JD, West MH, Sandbank T, Carter BJ. A human parvovirus, adeno-associated virus, as a eucaryotic vector: transient expression and encapsidation of the procaryotic gene for chloramphenicol acetyltransferase. Mol Cell Biol. 1984;4:2072–81.
Saraiva J, Nobre RJ, Pereira de Almeida L. Gene therapy for the CNS using AAVs: the impact of systemic delivery by AAV9. J Control Release. 2016;241:94–109.
Hocquemiller M, Giersch L, Audrain M, Parker S, Cartier N. Adeno-associated virus-based gene therapy for CNS diseases. Hum Gene Ther. 2016;27:478–96.
Naso MF, Tomkowicz B, Perry WL 3rd, Strohl WR. Adeno-associated virus (AAV) as a vector for gene therapy. BioDrugs. 2017;31:317–34.
Wang D, Tai PWL, Gao G. Adeno-associated virus vector as a platform for gene therapy delivery. Nat Rev Drug Discov. 2019;18:358–78.
Ke M-T, Fujimoto S, Imai T. SeeDB: a simple and morphology-preserving optical clearing agent for neuronal circuit reconstruction. Nat Neurosci. 2013;16:1154–61.
Tainaka K, Kubota SI, Suyama TQ, Susaki EA, Perrin D, Ukai-Tadenuma M, et al. Whole-body imaging with single-cell resolution by tissue decolorization. Cell. 2014;159:911–24.
Richardson DS, Lichtman JW. Clarifying tissue clearing. Cell. 2015;162:246–57.
Mano T, Albanese A, Dodt H-U, Erturk A, Gradinaru V, Treweek JB, et al. Whole-brain analysis of cells and circuits by tissue clearing and light-sheet microscopy. J Neurosci. 2018;38:9330 LP–9337. http://www.jneurosci.org/content/38/44/9330.abstract.
Hama H, Kurokawa H, Kawano H, Ando R, Shimogori T, Noda H, et al. Scale: a chemical approach for fluorescence imaging and reconstruction of transparent mouse brain. Nat Neurosci. 2011;14:1481–8.
Silvestri L, Costantini I, Sacconi L, Pavone FS. Clearing of fixed tissue: a review from a microscopist’s perspective. J Biomed Opt. 2016;21:81205.
Epp JR, Niibori Y, Liz Hsiang H-L, Mercaldo V, Deisseroth K, Josselyn SA, et al. Optimization of CLARITY for Clearing Whole-Brain and Other Intact Organs. eNeuro. 2015;2.
Isogai Y, Richardson DS, Dulac C, Bergan J. Optimized protocol for imaging cleared neural tissues using light microscopy. Methods Mol Biol. 2017;1538:137–53.
Erturk A, Becker K, Jahrling N, Mauch CP, Hojer CD, Egen JG, et al. Three-dimensional imaging of solvent-cleared organs using 3DISCO. Nat Protoc. 2012;7:1983–95.
Susaki EA, Tainaka K, Perrin D, Yukinaga H, Kuno A, Ueda HR. Advanced CUBIC protocols for whole-brain and whole-body clearing and imaging. Nat Protoc. 2015;10:1709–27.
Chen L, Li G, Li Y, Li Y, Zhu H, Tang L, et al. UbasM: An effective balanced optical clearing method for intact biomedical imaging. Sci Rep. 2017;7:12218.
Spalteholz W Über das Durchsichtigmachen von menschlichen und tierischen Präparaten, nebst Anhang: Über Knochenfärbung. Leipzig: S. Hirzel; 1911. 48 file://catalog.hathitrust.org/Record/009621299
Spalteholz W Über das Durchsichtigmachen von menschlichen und tierischen Präparaten und seine theoretischen Bedingungen: Nebst Anhang, Über Knochenfärbung. Leipzig: Verlag Von S. Hirzel; 1914.
Jensen KHR, Berg RW. Advances and perspectives in tissue clearing using CLARITY. J Chem Neuroanat. 2017;86:19–34.
Seo J, Choe M, Kim S-Y. Clearing and labeling techniques for large-scale biological tissues. Mol Cells. 2016;39:439–46.
Ueda HR, Ertürk A, Chung K, Gradinaru V, Chédotal A, Tomancak P, et al. Tissue clearing and its applications in neuroscience. Nat Rev Neurosci. 2020;21:61–79.
Tomer R, Ye L, Hsueh B, Deisseroth K. Advanced CLARITY for rapid and high-resolution imaging of intact tissues. Nat Protoc. 2014;9:1682–97.
Renier N, Wu Z, Simon DJ, Yang J, Ariel P, Tessier-Lavigne M. iDISCO: a simple, rapid method to immunolabel large tissue samples for volume imaging. Cell. 2014;159:896–910.
Bode J, Krüwel T, Tews B. Light sheet fluorescence microscopy combined with optical clearing methods as a novel imaging tool in biomedical research. Eur Med J. 2017;1:67–74.
Pan C, Cai R, Quacquarelli FP, Ghasemigharagoz A, Lourbopoulos A, Matryba P, et al. Shrinkage-mediated imaging of entire organs and organisms using uDISCO. Nat Methods. 2016;13:859–67.
Dodt H-U, Leischner U, Schierloh A, Jahrling N, Mauch CP, Deininger K, et al. Ultramicroscopy: three-dimensional visualization of neuronal networks in the whole mouse brain. Nat Methods. 2007;4:331–6.
Huisken J, Stainier DYR. Selective plane illumination microscopy techniques in developmental biology. Development. 2009;136:1963–75.
Park SH Composition for biotissue clearing and biotissue clearing method using same. Daejeon, KR; 2020. Available from: https://www.freepatentsonline.com/y2020/0271553.html.
Yates SC, Groeneboom NE, Coello C, Lichtenthaler SF, Kuhn P-H, Demuth H-U, et al. QUINT: workflow for quantification and spatial analysis of features in histological images from rodent brain. Front Neuroinform. 2019;13:75.
Groeneboom NE, Yates SC, Puchades MA, Bjaalie JG. Nutil: a pre- and post-processing toolbox for histological rodent brain section images. Front Neuroinform. 2020;14:37.
Puchades MA, Csucs G, Ledergerber D, Leergaard TB, Bjaalie JG. Spatial registration of serial microscopic brain images to three-dimensional reference atlases with the QuickNII tool. PLoS One. 2019;14:e0216796.
Berg S, Kutra D, Kroeger T, Straehle CN, Kausler BX, Haubold C, et al. ilastik: interactive machine learning for (bio)image analysis. Nat Methods. 2019;16:1226–32.
Zhang H, Yang B, Mu X, Ahmed SS, Su Q, He R, et al. Several rAAV vectors efficiently cross the blood-brain barrier and transduce neurons and astrocytes in the neonatal mouse central nervous system. Mol Ther. 2011;19:1440–8.
Fujishima K, Kawabata Galbraith K, Kengaku M. Dendritic self-avoidance and morphological development of cerebellar Purkinje cells. Cerebellum. 2018;17:701–8.
Kaneko M, Yamaguchi K, Eiraku M, Sato M, Takata N, Kiyohara Y, et al. Remodeling of monoplanar Purkinje cell dendrites during cerebellar circuit formation. PLoS One. 2011;6:e20108.
Fujishima K, Horie R, Mochizuki A, Kengaku M. Principles of branch dynamics governing shape characteristics of cerebellar Purkinje cell dendrites. Development. 2012;139:3442–55.
Nedelescu H, Abdelhack M, Pritchard AT. Regional differences in Purkinje cell morphology in the cerebellar vermis of male mice. J Neurosci Res. 2018;96:1476–89.
Sholl DA. Dendritic organization in the neurons of the visual and motor cortices of the cat. J Anat. 1953;87:387–406.
Lee E, Choi J, Jo Y, Kim JY, Jang YJ, Lee HM, et al. ACT-PRESTO: Rapid and consistent tissue clearing and labeling method for 3-dimensional (3D) imaging. Sci Rep. 2016;6:18631.
Kim S-Y, Cho JH, Murray E, Bakh N, Choi H, Ohn K, et al. Stochastic electrotransport selectively enhances the transport of highly electromobile molecules. Proc Natl Acad Sci USA. 2015;112:E6274–83.
Cai R, Pan C, Ghasemigharagoz A, Todorov MI, Förstera B, Zhao S, et al. Panoptic imaging of transparent mice reveals whole-body neuronal projections and skull–meninges connections. Nat Neurosci. 2019;22:317–27. https://doi.org/10.1038/s41593-018-0301-3.
Yang B, Treweek JB, Kulkarni RP, Deverman BE, Chen C-K, Lubeck E, et al. Single-cell phenotyping within transparent intact tissue through whole-body clearing. Cell. 2014;158:945–58.
Li W, Germain RN, Gerner MY. High-dimensional cell-level analysis of tissues with Ce3D multiplex volume imaging. Nat Protoc. 2019;14:1708–33.
Zaeck L, Potratz M, Freuling CM, Müller T, Finke S. High-resolution 3D imaging of rabies virus infection in solvent-cleared brain tissue. J Vis Exp. 2019. https://doi.org/10.3791/59402.
Hou B, Zhang D, Zhao S, Wei M, Yang Z, Wang S, et al. Scalable and DiI-compatible optical clearance of the mammalian brain. Front Neuroanat. 2015;9:19.
Hama H, Hioki H, Namiki K, Hoshida T, Kurokawa H, Ishidate F, et al. ScaleS: an optical clearing palette for biological imaging. Nat Neurosci. 2015;18:1518–29.
Qi Y, Yu T, Xu J, Wan P, Ma Y, Zhu J, et al. FDISCO: Advanced solvent-based clearing method for imaging whole organs. Sci Adv. 2019;5:eaau8355.
Schwarz MK, Scherbarth A, Sprengel R, Engelhardt J, Theer P, Giese G. Fluorescent-protein stabilization and high-resolution imaging of cleared, intact mouse brains. PLoS One. 2015;10:e0124650 https://doi.org/10.1371/journal.pone.0124650.
Karagiannis ED, Boyden ES. Expansion microscopy: development and neuroscience applications. Curr Opin Neurobiol. 2018;50:56–63.
Gombash Lampe SE, Kaspar BK, Foust KD. Intravenous injections in neonatal mice. J Vis Exp. 2014;e52037. https://doi.org/10.3791/52037.
Hirai H. Progress in transduction of cerebellar Purkinje cells in vivo using viral vectors. Cerebellum. 2008;7:273–8.
Cook AA, Fields E, Watt AJ. Losing the beat: contribution of purkinje cell firing dysfunction to disease, and its reversal. Neuroscience. 2021;462:247–61.
Mavroudis IA, Petrides F, Manani M, Chatzinikolaou F, Ciobică AS, Pădurariu M, et al. Purkinje cells pathology in schizophrenia. A morphometric approach. Rom J Morphol Embryol. 2017;58:419–24.
Erturk A, Mauch CP, Hellal F, Forstner F, Keck T, Becker K, et al. Three-dimensional imaging of the unsectioned adult spinal cord to assess axon regeneration and glial responses after injury. Nat Med. 2011;18:166–71.
Jing D, Zhang S, Luo W, Gao X, Men Y, Ma C, et al. Tissue clearing of both hard and soft tissue organs with the PEGASOS method. Cell Res. 2018;28:803–18.
Chiang A-S, Lin W-Y, Liu H-P, Pszczolkowski MA, Fu T-F, Chiu S-L, et al. Insect NMDA receptors mediate juvenile hormone biosynthesis. Proc Natl Acad Sci USA. 2002;99:37–42.
Staudt T, Lang MC, Medda R, Engelhardt J, Hell SW. 2,2’-thiodiethanol: a new water soluble mounting medium for high resolution optical microscopy. Microsc Res Tech. 2007;70:1–9.
Aoyagi Y, Kawakami R, Osanai H, Hibi T, Nemoto T. A rapid optical clearing protocol using 2,2′-thiodiethanol for microscopic observation of fixed mouse brain. PLoS One. 2015;10:e0116280. https://doi.org/10.1371/journal.pone.0116280.
Costantini I, Ghobril J-P, Di Giovanna AP, Allegra Mascaro AL, Silvestri L, Mullenbroich MC, et al. A versatile clearing agent for multi-modal brain imaging. Sci Rep. 2015;5:9808.
Tsai PS, Kaufhold JP, Blinder P, Friedman B, Drew PJ, Karten HJ, et al. Correlations of neuronal and microvascular densities in murine cortex revealed by direct counting and colocalization of nuclei and vessels. J Neurosci. 2009;29:14553–70.
Kuwajima T, Sitko AA, Bhansali P, Jurgens C, Guido W, Mason C. ClearT: a detergent- and solvent-free clearing method for neuronal and non-neuronal tissue. Development. 2013;140:1364–8.
Li W, Germain RN, Gerner MY. Multiplex, quantitative cellular analysis in large tissue volumes with clearing-enhanced 3D microscopy (Ce3D). Proc Natl Acad Sci U S A. 2017;114:E7321–30.
Susaki EA, Tainaka K, Perrin D, Kishino F, Tawara T, Watanabe TM, et al. Whole-brain imaging with single-cell resolution using chemical cocktails and computational analysis. Cell. 2014;157:726–39.
Chung K, Wallace J, Kim S-Y, Kalyanasundaram S, Andalman AS, Davidson TJ, et al. Structural and molecular interrogation of intact biological systems. Nature. 2013;497:332–7.
Murray E, Cho JH, Goodwin D, Ku T, Swaney J, Kim S-Y, et al. Simple, scalable proteomic imaging for high-dimensional profiling of intact systems. Cell. 2015;163:1500–14.
Funding
This work was funded by the ERDF through the Regional Operational Program Center 2020, Competitiveness Factors Operational Program (COMPETE 2020) and National Funds through FCT (Foundation for Science and Technology): Imagene (PTDC/BBB-NAN/0932/2014 | POCI-01-0145-FEDER-016807), MODELPOLYQ 2.O (CENTRO-01-0145-FEDER-181258), MJDEDIT (CENTRO-01-0145-FEDER-181266), BDFORMJD (CENTRO-01-0145-FEDER-181240), CENTRO-01-0246-FEDER-000010 (Multidisciplinary Institute of Ageing in Coimbra), BrainHealth2020 projects (CENTRO-01-0145-FEDER-000008), UID/NEU/04539/2019, UIDB/04539/2020, UIDP/04539/2020, LA/P/0058/2020, PPBI (POCI-01-0145-FEDER-022122), ViraVector (CENTRO-01-0145-FEDER-022095), CortaCAGs (PTDC/NEU-NMC/0084/2014 | POCI-01-0145-FEDER-016719), SpreadSilencing (POCI-01-0145-FEDER-029716), CancelStem (POCI-01-0145-FEDER-016390), POCI-01-0145-FEDER-030737, POCI-01-0145-FEDER-032309, as well as SynSpread, ESMI and ModelPolyQ under the EU Joint Program – Neurodegenerative Disease Research (JPND), the last two co-funded by the European Union H2020 program, GA No.643417; by National Ataxia Foundation (USA), the American Portuguese Biomedical Research Fund (APBRF) and the Richard Chin and Lily Lock Machado-Joseph Disease Research Fund. MML was supported by a PhD fellowship from FCT (2021.05776.BD).
Author information
Authors and Affiliations
Contributions
Conceptualization, MML, LPA, LC, RJN; Methodology, MML, LC, RJN; Investigation, MML, JP, JR, SML, LC; Formal Analysis, MML, LC; Visualization, MML, SML, LC; Writing – Original Draft, MML; Writing – Review & Editing, MML, JP, JR, SML, LPA, LC, RJN; Resources, JP, JR, LPA; Funding Acquisition, LPA, LC, RJN; Supervision, LPA, LC, RJN.
Corresponding authors
Ethics declarations
Competing interests
The authors received no specific funding from Binaree, Inc. for the development of the present work. JP works for Carl Zeiss Microscopy GmbH, ZEISS Group. The other authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Lopes, M.M., Paysan, J., Rino, J. et al. A new protocol for whole-brain biodistribution analysis of AAVs by tissue clearing, light-sheet microscopy and semi-automated spatial quantification. Gene Ther 29, 665–679 (2022). https://doi.org/10.1038/s41434-022-00372-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41434-022-00372-z
