
Abstract
Previously, we developed a chimeric adenovirus type 5 with type 35 fiber (Ad5/35), which has high tropism to dendritic cells and low hepatoxicity. For further clinical use, we constructed two recombinant vectors expressing human immunodeficiency virus 1 (HIV-1) clade C gag (Ad5/35-Cgag and MVA-Cgag). The biodistribution of the two viral vectors in a mouse model and immunity in monkeys were assessed. The mice received a single intramuscular injection with the vectors alone. The gag gene in the tissues were periodically detected using a real-time quantitative polymerase chain reaction. The distribution of Ad5/35 was also detected using an in vivo imaging system, followed by luciferase-expressing Ad5/35 administration. We found that Ad5/35-Cgag DNA and luciferase activity were detectable until 8 weeks post-administration, whereas MVA-Cgag was undetectable 72 h post-administration. Furthermore, viral administration did not increase serum aspartate aminotransferase and alanine aminotransferase levels in either mouse or monkey models. Moreover, intramuscular administration of Ad5/35-Cgag induced the gag-specific antibody level and IFNγ-secreting PBMCs, the boost with MVA-Cgag further increased the responses and lasted more than 20 weeks from the initial administration. These data demonstrate that Ad5/35 and MVA vectors are safe for in vivo use, and prime-boost with Ad5/35-MVA vaccines is suitable for clinical use against HIV-1 clade C.
Similar content being viewed by others

Introduction
In 2019, 38 million people worldwide were infected with the human immunodeficiency virus (HIV), with 1.7 million new infections and 690,000 HIV-related deaths (https://aids2020.unaids.org). A third of the world’s new infections occur in southern Africa, where HIV-1 clade C predominates [1]. Despite drastic improvements in access to antiretroviral therapy (ART), only approximately 59% of people living with HIV have thus far received ART. Furthermore, once the treatment begins, a lifetime of multidrug therapy is required [2]. Few acquired immunodeficiency syndrome drugs have serious side effects, and patients may have to stop taking them because of these effects. The emergence of drug-resistant viruses is also a major problem, with 20–40% of patients who start multidrug therapy reporting poor outcomes (https://aids2020.unaids.org). Although several vaccines against HIV have been explored in clinical trials, they do not induce effective protection against HIV infection [3]. Therefore, it would be both practical and beneficial to develop an HIV-1 vaccine that can prevent infection, even if partially.
To date, several vaccine candidates have completed clinical efficacy trials. According to the VaxGen (envelope [env] gp120 protein vaccine, n = 5108) [4] and HVTN 505 (DNA vaccine prime/adenovirus type 5 (Ad5) vector vaccine boost, n = 2504) [5] studies, both vaccines failed to protect against infection or failed to reduce viral loads after HIV-1 infection [6]. A large phase three trial (study ID: RV144, n = 16,402) of a recombinant canary pox vector prime with a gp120 protein boost was completed in Thailand as of September 2009, with an efficacy announcement that this HIV vaccine regimen was safe and moderately effective at reducing the rate of HIV infection compared to those were the placebo [7]. This study is the first evidence that a prime-boost HIV vaccine regimen may prevent infection and represents a significant step forward in HIV vaccine research. However, the same protocol (study ID: HVTN 702, n = 5407) in South Africa did not show any significant protective efficacy [8]. Another study (study ID: HVTN 705, n = 2637) using Ad26 vector vaccine prime/env gp140 protein vaccine boost did not provide sufficient protection against HIV infection in a population of young women in sub-Saharan Africa at high risk of acquiring HIV (https://www.prnewswire.com).
Given the results of the past studies on HIV vaccines, the prime-boost regimen is able to induce much stronger immunity than that immunization with a single vaccine. Viral vector-based vaccines have shown promising results [9,10,11]. Among these vectors, replication-defective Ad5 recombinants (with deletions affecting the essential replication genes E1 and E3) and a replication-defective modified vaccinia Ankara virus (MVA) elicited the most potent cellular immunity and provided the highest degree of protection in non-human primates [10, 11]. A major limitation for the clinical application of Ad5 is the pre-existing immunity against Ad5 fiber and hexon in humans and Ad5-related hepatoxicity [12,13,14]. To overcome these shortcomings, we constructed an Ad5-based chimeric vector containing Ad type 35 fiber (Ad5/35), which binds to the CD46 receptor for cell entry and can avoid some of the pre-existing Ad5 antibody [15]. Previously, using a regimen with Ad5/35 vaccine primer/MVA vaccine boost in a non-primate animal model, both partial protection from the intravenous challenge of a high pathogenic strain (SIVmac239) [16] and a significant therapeutic efficacy [17] were obtained.
For further clinical application, in this study, we constructed an HIV-1 clade C gag-expressing Ad5/35 vector (Ad5/35-Cgag) and an MVA vector (MVA-Cgag). Biodistribution and immunity were assessed in a mouse model and non-human primates, respectively.
Results
Specificity of RT-qPCR in detecting the gag gene of Ad5/35 and MVA vectors
To assess the specificity of real-time quantitative polymerase chain reaction (RT-qPCR), we added Ad5/35-Cgag and MVA-Cgag genomic DNA to whole blood genomic DNA extracted from monkeys, mice, and rats followed by RT-qPCR. We found that the gag gene (Supplementary Fig. S1) could be detected even in the presence of an excess amount of whole blood genomic DNA. No specific amplification was observed from the respective DNA, confirming the specificity of the primers used for gag gene detection.
To quantify the gag gene, we mixed series copy numbers of Ad5/35-Cgag and MVA-Cgag genomic DNA with genomic DNA extracted from various mouse tissues (brain, kidney, liver, muscle, lung, heart, intestinal lymph nodes, spleen, thymus, gonads, and blood), followed by RT-qPCR. We found that target DNA concentrations from 10–1 × 106 copies/μL showed good linearity over the entire quantitative range for the gag gene (Supplementary Fig. S2). All correlation coefficients (r) were > 0.99.
Biodistribution of Ad5/35-Cgag and MVA-Cgag in vivo
To assess the distribution of Ad and MVA vectors, BALB/c mice were intramuscularly injected with 2 × 109 viral particles (vp) of Ad-Cgag, and the gag gene in the tissues were examined over time. The Ad copy number detected in the muscle was significantly higher than that in other tissues at 1 week after Ad5/35-Cgag administration (Table 1 and Fig. 1). The Ad copy number detected in the muscle was significantly higher than that in other tissues up to 4 weeks after administration and decreased over time to a level that was no longer significantly different from that in other tissues after 8 weeks of administration. Besides the injection site, a lower copy number was detected in other tissues, suggesting that it may have leaked from the injection site following blood flow.

BALB/c mice (n = 5) were intramuscularly injected with Ad5/35-Cgag, and the gag gene was quantified using real-time PCR in each tissue at the indicated time points. The bars represent means ± SE. *p < 0.05; **p < 0.01; ***p < 0.001.
Similarly, 2 × 107 plaque-forming units (pfu) MVA-Cgag was intramuscularly injected, and tissue distribution was examined over time. Compared to Ad5/35-Cgag detected at 8 weeks post-administration, MVA-Cgag was not detected 72 h after vector injection (Table 2 and Fig. 2). The vector was also detectable in other tissues with low copy numbers. These results indicate that MVA may induce much higher innate and/or cellular immunity against vector than Ad5/35, which results in the different existing periods in vivo.

BALB/c mice (n = 5) were intramuscularly injected with MVA-Cgag, and the gag gene was quantified using real-time PCR in each tissue at the indicated time points. The bars represent means ± SE. *p < 0.05; **p < 0.01; ***p < 0.001.
Biodistribution of Ad5/35-Luc in vivo
Next, to further investigate the biodistribution of the Ad vector in vivo, BALB/c mice were intramuscularly injected with luciferase-expressing Ad5/35 vector (Ad5/35-Luc) and monitored over time using an in vivo imaging system (IVIS). Luciferase expression was primarily observed at the injection site, and the fluorescence that appeared on day 1 peaked on day 7, then decreased over time and disappeared on day 28 (Fig. 3). A low fluorescence was observed in the other organs including the liver. As a control, the same dose of Ad5-Luc was intramuscularly injected to BALB/c mice and Luciferase activity was detected by IVIS on day 7 post-administration. As shown in Fig. 3, higher luciferase activity was detected in the liver compared to the mice administered with Ad5/35-Luc. These data confirmed our RT-qPCR results and suggested that the chimeric adenovirus Ad5/35 can be used as a vaccine with a lower risk of hepatotoxicity.

BALB/c mice (n = 5) were intramuscularly injected with Ad5/35-Luc. Luciferase activity in vivo was detected using an IVIS at the indicated time points (left). Bioluminescence was measured and expressed as the mean ± SE (right). As a control, the BALB/c mouse was intramuscularly injected with Ad5-Luc. Luciferase activity was detected using an IVIS on day 7.
Hepatotoxicity post-Ad5/35 or MVA vector administration
To explore whether hepatotoxicity occurred after viral vector administration, the mice were intramuscularly injected with 2 × 109 vp Ad5/35-Cgag or 2 × 107 pfu of MVA-Cgag. A 2 × 109 vp Ad5-Cgag was used as the positive control. Serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels were measured on days 0, 3, 7, 14, and 28. As shown in Fig. 4A, B, the ALT and AST levels were not different from those of pre-injection, post-Ad5/35-Cgag, or MVA-Cgag administration, but high ALT and AST levels were found in Ad5-administrated mice.

BALB/c mice (A, B) were intramuscularly injected with Ad5/35-Cgag (n = 5), Ad5-Cgag (n = 3), or MVA-Cgag (n = 5). Rhesus monkeys (n = 3) were intramuscularly injected with Ad5/35-Cgag and MVA-Cgag at weeks 0 and 8, respectively. Rhesus monkeys (n = 2) were intramuscularly injected with Ad5/35-GFP and MVA-GFP at weeks 0 and 8, respectively. C, D Serum AST and ALT levels were detected at the indicated time points.
We then explored whether Ad5/35 prime/MVA boost-induced hepatotoxicity in monkeys. The vaccine group (n = 3) was intramuscularly administered 1011 vp of Ad5/35-Cgag and 108 pfu of MVA-Cgag at weeks 0 and 8, respectively. The control group (n = 2) administrated the same dose of Ad5/35-GFP and MVA-GFP. Serum ALT and AST levels were measured at weeks 0, 2, 4, 8, 10, and 12. As shown in Fig. 4C, D, the ALT and AST levels were not different from those of the pre-injection and post-administration of Ad5/35 and MVA vectors in rhesus monkeys.
Immune response in rhesus monkeys after vaccination
Prime immunization with Ad5/35-Cgag induced gag-specific serum IgG to 11–12 log2 and boost immunization with MVA-Cgag further increased the antibody titer to 13–16 log2 (Fig. 5A). A high titer was observed until 3 months after boost immunization.

Monkeys were intramuscularly administrated as follows: HIV vaccine group (n = 3), Ad5/35-Cgag and MVA-Cgag; Control group (n = 2), Ad5/35-GFP and MVA-GFP. HIV Cgag-specific serum IgG titer (A) and HIV Cgag-specific IFNγ cells (B) were detected.
Next, we explored the cellular immune responses with a prime-boost regimen. Immunization with Ad5/35-Cgag increased gag-specific IFNγ-secreting cells to 750–970 per million PBMCs. Boost immunization with MVA-Cgag further increased the cells to 2600–3300 per million PBMCs. These data demonstrate that the prime-boost regimen can further increase both humoral and cellular immune responses.
Discussion
The aim of this study was to evaluate the efficacy of vaccination methods by examining the biodistribution of the viral vaccine and immune responses to HIV-1 through vaccination with a prime-boost regimen using chimeric Ad5/35-Cgag and MVA-Cgag. The results of the present study showed that Ad5/35-Cgag and MVA-Cgag are present at the site of administration for a certain period of time with limited diffusion to other tissues, and no serious hepatoxicity after viral administration was observed, along with the production of high anti-gag antibodies and cellular immunity. Furthermore, boosting MVA-Cgag enhances both humoral and cellular immune responses induced by Ad5/35-Cgag.
Viral vectors based on human adenovirus types 5 and 26 have been used as prophylactic HIV vaccines in phase II clinical trials [18]. Severe acute respiratory syndrome coronavirus 2 (SARS-Cov-2) vaccines based on Ad5, Ad26, and chimpanzee adenovirus (ChAdOx1) have been approved for emergency use in humans [19, 20]. However, in very few cases, thrombosis and thrombocytopenia have been reported in vaccinees who received Ad26 or ChAdOx1-based SARS-CoV-2 vaccines [21,22,23]. More attention has been paid to the safety of adenovirus vectors.
In this study, the biodistribution of Ad5/35-Cgag by RT-qPCR showed that it was mainly present in the muscle at the injection site, starting from 1 week after injection and gradually decreasing until 8 weeks (Fig. 1 and Table 1). The relatively long period of immunogen expression may induce a longer and stronger immune response. However, this also raises the safety issue of adenovirus vectors. Similar to Ad5/35-Cgag, MVA-Cgag was primarily detected at the injection site using RT-qPCR, but viral localization was shorter than that of Ad5/35-Cgag (Fig. 2 and Table 2). Imaging with IVIS was consistent with this result, and expression of luciferase by Ad5/35-Luc was observed at the injection site from day 1 to 4 weeks, peaking at 1 week after administration into the muscle (Fig. 3). Since it has been reported that luciferase expression by MVA-Luc becomes undetectable 48 h after administration [24], the short length of localization may be due to the strong activation of innate immunity by MVA [25]. A study of the distribution of viral vectors by Hank et al. showed that MVA vectors are not toxic to various organs [26]. Clinical safety studies in healthy HIV-1-infected volunteers have shown similar results [27]. However, the short localization of MVA in vivo is not considered a major hindrance in terms of vaccine application. The short virus disappearance time indicates that there is little risk of virus persistence in vivo, and multiple boosts, if necessary, would be sufficient to achieve vaccine efficacy. In fact, in our prime-boost regimen, a single boost of MVA-Cgag enhanced immune responses by Ad5/35-Cgag (Fig. 5).
Regarding viral distribution, most studies administered viruses via intravenous routes [28, 29]. In this study, we explored the distribution of Ad5/35-Cgag and MVA-Cgag via the intramuscular route, which is a popular immunization route for most vaccines used in humans. In this study, Ad5/35 and MVA vectors were intramuscularly administered to mice and rhesus monkeys. RT-qPCR analysis and IVIS showed that the viral vaccine was largely retained at the injection site, with minimal diffusion into the other tissues (Figs. 1–3). Therefore, the use of Ad5/35 as a vaccine vector is expected to have a lower risk of hepatotoxicity compared to that with the use of the Ad5 vector, which is known to exhibit hepatotoxicity as a side effect [14]. It has been reported that Ad5/35 infects hepatic parenchymal cells at level 4–5 log orders lower than that of Ad5, and Ad5/35 cannot infect non-hepatic parenchymal cells [9, 15]. Intramuscular administration of the Ad3/35 or MVA vector did not affect serum ALT and AST levels in either mouse and or monkeys (Fig. 4). Other side effects were also previously reported, such as infection of the central nervous system by intranasal administration of the Ad5 vector [30] and fatal encephalitis in immunosuppressed HIV-infected individuals caused by vaccinia virus [31]. The spread of Ad5/35-Cgag and MVA-Cgag to the brain was not observed in this study.
The prime-boost regimen used for rhesus monkeys in this study was based on the results of a detailed analysis of viral vaccine biodistribution and maintenance in mice (Figs. 1–3). The RT-qPCR analysis performed in this study to detect HIV gag was able to detect DNA extracted from tissue samples at a concentration as low as 10 copies/μL (equivalent to 50 copies per PCR reaction; Supplementary Fig. S2). The standard curve for 6-digit DNA concentrations (10–106 copies/μL) generated using PCR showed good linearity. Assay sensitivity was assessed by repeating the amplification of the lowest concentration (10 copies/μL) in a background of 1 μg genomic DNA for a total of five points on the standard curve. The RSD of each assay was sufficiently low to guarantee the stability and reliability of the evaluation. The recovery rate of MVA from blood was lower than that of other tissues, which was low as described elsewhere [32], but this observation may be due to the fact that nucleic acids in blood are often contaminated with components that inhibit PCR [33].
Vaccines based on prime-boost regimens have been studied in a variety of proteins, DNA vaccines, and viral vectors, but often require multiple doses to induce a strong immune response [16, 34, 35]. In this study, to evaluate the constructed Ad5/35-Cgag and MVA-Cgag as a vaccine in a basic way, we decided to administer only one boost to rhesus monkeys. One week after intramuscular administration of Ad5/35-Cgag, the production of anti-gag humoral and cellular immunity was observed, and the production peaked after 6 weeks (Fig. 5). When MVA-Cgag was administered 8 weeks after the initial immunization, the gag-specific immune responses by Ad5/35-Cgag were enhanced. This indicates that a prime-boost regimen of HIV vaccine using chimeric Ad and recombinant MVA as vectors is useful in non-human primates.
Despite the extensive development of HIV vaccines, many problems remain unsolved. One of them is the lack of animal models that can faithfully reproduce human immune responses to HIV. For example, the rapid progression of the disease induced by the simian HIV SHIV89.6 P is very different from that of HIV [36]. Furthermore, many experiments in non-human primates have been challenged by the intravenous route, which is different from most natural infections via the mucosal route [16, 35]. Thus, HIV research remains a formidable scientific and public health challenge, and further effort is urgently required to achieve the ultimate goal of a safe and effective HIV-1 vaccine.
In conclusion, this study assessed the biodistribution of Ad5/35-Cgag and MVA-Cgag in mice and the immune responses of a prime-boost regimen with the vaccines in non-human primates. We found that the two vaccines were safe and induced strong and periodic immune responses. Thus, prime-boost with Ad5/35-Cgag and MVA-Cgag may be highly suitable for clinical use.
Materials and methods
Animals and ethics statement
BALB/c mice (20–25 g) were purchased from the Experimental Animal Center at the Academy of Military Medical Science of PLA (Beijing, China). Rhesus macaques (approximately 4.0 kg) were purchased from Xie’e’xin Experimental Animal Center (Beijing, China). The experiments were performed at the Experimental Animal Center at Beijing Institute of Microbiology and Epidemiology. Both the mice and primates were maintained in pathogen-free environments. All experimental procedures were carried out in accordance with the protocols of the Administrative Panel on Laboratory Animal Care and the guidelines set by the Beijing Institute of Microbiology and Epidemiology. The protocols used in this study were approved by the Institute Animal Care and Use Committee/Ethics Committee of Beijing Institute of Microbiology and Epidemiology.
Recombinant vectors
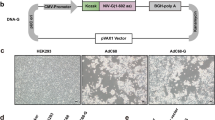
An E1/E3-deletion, replication-defective chimeric Ad5 vector with the Ad 35 fiber expressing the codon-optimized full-length gag gene from HIV-1 subtype C (strain 96ZM651.8, GenBank accession no. Af286224; A5/35-Cgag) was constructed as previously described [37]. The Ad vector was propagated in human embryonic kidney 293 cells and purified using CsCl gradient centrifugation. The total concentration of virions was calculated using the following formula: optical density at 260 nm (OD260) = 1012 viral particles (vp).
The expression cassette was subcloned into a replication-defective recombinant MVA vector to generate recombinant MVA-Cgag, as previously described [16, 17, 38]. MVA-Cgag virus was propagated in BHK21 cells, purified using one round of ultra-centrifugation over 36% sucrose, and titrated into BHK21 cells to determine the number of plaque-forming units (pfu).
Biodistribution of A5/35-Cgag and MVA-Cgag in BALB/c mice
The A5/35-Cgag vector (100 μL) containing 2 × 109 vp was injected intramuscularly into the left femoral muscles of BALB/c mice. Five mice were used at each time point. At weeks 1, 2, 4, and 8, the mice were sacrificed, and the blood and tissue samples (heart, liver, spleen, lung, kidney, brain, mesenteric lymph node, gonads, muscle, and thymus) were collected for subsequent analysis. An E.Z.N.A. Tissue DNA kit and E.Z.N.A. Blood DNA Mini Kit (Omega, Norcross, GA, USA) were used for the extraction of genomic DNA from tissue and blood, respectively, following the manufacturer’s instructions. The samples were also collected at 0.5, 8, 24, and 72 h after intramuscular injection of 2 × 107 pfu MVA-Cgag vector. The samples were immediately frozen by immersion in liquid nitrogen and stored at –70 °C until further analysis.
Serum ALT and AST were detected using DRI-CHEM NX500V with Dry-Chem Slides for GOT/AST-PIII and GFP/ALT-PIII (Fujifilm, Tokyo, Japan) on days 0, 3, 7, 14, and 28 for mice and at weeks 0, 2, 4, 8, 10, and 12 for monkeys after intramuscular administration. Intramuscular injection of 2 × 109 vp Ad5-Cgag was used as a positive control.
RT-qPCR assay for mouse tissues
Primers and TaqMan probes were selected using Primer Premier 5.0, and Primer Express 2.0. The primer pairs (Forward: 5’-CTACAAGCGCTGGATCATCCT-3’; Reverse: 5’-TTGAAGAAGCGGTCCACGTAGT-3’) and probe (FAM-ACAAGATCGTGCGCATGTACAGCCC-TAMRA) were used for detection of the HIV gag gene. Protocol optimization was performed by evaluating the effects of a range of primer and probe concentrations. For negative control, at least one no-template control (NTC; reactions that contained all PCR components except template DNA) was included in each PCR run to confirm the absence of both template and product contamination in the PCR mixture [39, 40]. Amplification was performed in a 20 μL reaction system as previously described [41]. Data analysis was performed using LightCycler Software 4.05 (Roche Life Science, Penzburg, Germany).
In vivo bioluminescence measurement of Ad5/35-Luc
The investigation was performed as previously described [42]. BALB/c mice (n = 5) were intramuscularly injected with 2 × 109 vp of the Ad5/35-Luc vector in 100 μL physiological saline. On days 1, 3, 7, 10, 14, 21, and 28, the mice were anesthetized with 2% pentobarbital sodium, and a single dose of 150 mg/kg luciferin in 100 μL of normal saline was intraperitoneally administered. Twenty minutes after injection of luciferin, charge-coupled device (CCD) images were obtained using a cooled IVIS CCD camera (Berthold Technologies, Bad Wildbad, Germany) with an integration time of 30 s and analyzed. The same dose of Ad5-Luc vector was used as a control and luciferase activity was detected on day 7 after administration.
Immunization in monkeys
To explore the immunogenicity of the viral vaccines in monkeys, three rhesus macaques were intramuscularly immunized with 1011 vp of Ad5/35-Cgag and boosted with 108 pfu MVA-Cgag at week 8. In the control group, two rhesus macaques were intramuscularly injected with 1011 vp of the Ad5/35-GFP vector and 108 pfu of the MVA-GFP vector on day 0.
Blood samples were drawn from the monkeys at weeks 0, 2, 4, 8, 10, 12, 14, 16, and 20, and centrifuged at 2000 g for 10 min to separate the serum from the blood cells. The sera were stored at –70 °C until further use.
Enzyme-linked immunosorbent assay (ELISA)
The HIV-1 clade C p24 protein was expressed in Escherichia coli using a pGEX6p-1 plasmid vector and purified using a GSTrap FF affinity column, as previously described in a study by our laboratory [43]. The HIV-1-specific antibody was detected using an ELISA [16, 37]. In brief, 96-well microtiter plates were coated with 10 μg/mL of HIV-1 clade C p24 protein and incubated overnight at 4 °C. The wells were blocked with phosphate-buffered saline containing 1% bovine serum albumin for 2 h at 37 °C. They were then treated with 100 μL of antisera and incubated for an additional 2 h at 37 °C. The bound immunoglobulin was quantified using an affinity-purified horseradish peroxidase-labeled anti-monkey IgG (Alpha Diagnostics, San Antonio, TX, USA). Absorbance was measured at a wavelength of 450 nm using a microplate reader (Model 550; Bio-Rad, Hercules, CA, USA). Endpoint titers were computed as the reciprocal of the highest dilution that had an absorbance value > 0.2 after the background subtraction (absorbance of the wells lacking a sample). Values higher than or equal to a two-fold rise after vaccination were plotted.
ELISPOT assay
The frequency of HIV-specific IFN-γ-secreting cells in monkeys was determined using an ELISPOT assay kit (U-Cytech, Utrech, The Netherlands) according to the manufacturer’s manual. In brief, 2 × 105 monkey PBMCs were stimulated in triplicate wells with 10 μg/mL of 15-mer animo acid peptide pool with 11-mer overlap of HIV consensus subtype C gag (obtained from AIDS Research and Reference Reagent Program) for 16 h at 37 °C. Non-stimulated cells were used to assess the background. The cells were transferred to an anti-IFN-γ Ab-coated 96-well plate and incubated for 5 h at 371 C. The cells were removed and 200 μL/well of ice-cold deionized water was added to lyse the remaining PBMCs. Subsequently, the plate was washed with PBS containing 0.05% Tween 20 (PBS-T) and incubated with biotinylated anti-IFN-γ Ab for 1 h at 37 °C. After 10 washings with PBS-T, 50 μl of gold-labeled anti-biotin Ab was added and incubated for 1 h at 37 oC. The plate was washed 10 times with PBS-T, and 30 μL of activator solutions were added. The plate was incubated in the dark for 30 min at room temperature to develop spot formations. After 30 min incubation, the plate was washed with deionized water and air-dried; spots were counted by a computer-assisted video image analysis. The results were expressed as spot-forming cells (SFC) per million cells.
Data analysis
All values are expressed as means ± SE. Statistical analysis of the experimental and control data was conducted using a one-way factorial analysis of variance (ANOVA). For all statistical analyses, significance was defined as P < 0.05.
References
Bbosa N, Kaleebu P, Ssemwanga D. HIV subtype diversity worldwide. Curr Opin HIV AIDS. 2019;14:153–60.
Bandera A, Gori A, Clerici M, Sironi M. Phylogenies in ART: HIV reservoirs, HIV latency and drug resistance. Curr Opin Pharmacol. 2019;48:24–32.
Pitisuttithum P, Marovich MA. Prophylactic HIV vaccine: vaccine regimens in clinical trials and potential challenges. Expert Rev Vaccines. 2020;19:133–142.
Pitisuttithum P, Gilbert P, Gurwith M, Heyward W, Martin M, van Griensven F, et al. Randomized, double-blind, placebo-controlled efficacy trial of a bivalent recombinant glycoprotein 120 HIV-1 vaccine among injection drug users in Bangkok, Thailand. J Infect Dis. 2006;194:1661–71.
Hammer SM, Sobieszczyk ME, Janes H, Karuna ST, Mulligan MJ, Grove D, et al. Efficacy trial of a DNA/rAd5 HIV-1 preventive vaccine. N Engl J Med. 2013;369:2083–92.
Barouch DH. Challenges in the development of an HIV-1 vaccine. Nature. 2008;455:613–9.
Rerks-Ngarm S, Pitisuttithum P, Nitayaphan S, Kaewkungwal J, Chiu J, Paris R, et al. Vaccination with ALVAC and AIDSVAX to prevent HIV-1 infection in Thailand. N Engl J Med. 2009;361:2209–20.
Gray GE, Bekker LG, Laher F, Malahleha M, Allen M, Moodie Z, et al. Vaccine efficacy of ALVAC-HIV and bivalent subtype C gp120-MF59 in adults. N Engl J Med. 2021;384:1089–1100.
Xin KQ, Jounai N, Someya K, Honma K, Mizuguchi H, Naganawa S, et al. Prime-boost vaccination with plasmid DNA and a chimeric adenovirus type 5 vector with type 35 fiber induces protective immunity against HIV. Gene Ther. 2005;12:1769–77.
Shiver JW, Fu TM, Chen L, Casimiro DR, Davies ME, Evans RK, et al. Replication-incompetent adenoviral vaccine vector elicits effective anti-immunodeficiency-virus immunity. Nature. 2002;415:331–5.
Amara RR, Villinger F, Staprans SI, Altman JD, Montefiori DC, Kozyr NL, et al. Different patterns of immune responses but similar control of a simian-human immunodeficiency virus 89.6P mucosal challenge by modified vaccinia virus Ankara (MVA) and DNA/MVA vaccines. J Virol. 2002;76:7625–31.
Roelvink PW, Lizonova A, Lee JG, Li Y, Bergelson JM, Finberg RW, et al. The coxsackievirus-adenovirus receptor protein can function as a cellular attachment protein for adenovirus serotypes from subgroups A, C, D, E, and F. J Virol. 1998;72:7909–15.
Adams JY, Johnson M, Sato M, Berger F, Gambhir SS, Carey M, et al. Visualization of advanced human prostate cancer lesions in living mice by a targeted gene transfer vector and optical imaging. Nat Med. 2002;8:891–7.
Thomas CE, Ehrhardt A, Kay MA. Progress and problems with the use of viral vectors for gene therapy. Nat Rev Genet. 2003;4:346–358.
Sakurai F, Mizuguchi H, Yamaguchi T, Hayakawa T. Characterization of in vitro and in vivo gene transfer properties of adenovirus serotype 35 vector. Mol Ther. 2003;8:813–21.
Wang HB, Kondo A, Yoshida A, Yoshizaki S, Abe S, Bao LL, et al. Partial protection against SIV challenge by vaccination of adenovirus and MVA vectors in rhesus monkeys. Gene Ther. 2010;17:4–13.
Shimada M, Wang HB, Kondo A, Xu XP, Yoshida A, Shinoda K, et al. Effect of therapeutic immunization using Ad5/35 and MVA vectors on SIV infection of rhesus monkeys undergoing antiretroviral therapy. Gene Ther. 2009;16:218–28.
Laher F, Bekker LG, Garrett N, Lazarus EM, Gray GE. Review of preventative HIV vaccine clinical trials in South Africa. Arch Virol. 2020;165:2439–2452.
Zhao J, Zhao S, Ou J, Zhang J, Lan W, Guan W, et al. COVID-19: coronavirus vaccine development updates. Front Immunol. 2020;11:602256.
Ura T, Yamashita A, Mizuki N, Okuda K, Shimada M. New vaccine production platforms used in developing SARS-CoV-2 vaccine candidates. Vaccine. 2021;39:197–201.
Bayas A, Menacher M, Christ M, Behrens L, Rank A, Naumann M. Bilateral superior ophthalmic vein thrombosis, ischaemic stroke, and immune thrombocytopenia after ChAdOx1 nCoV-19 vaccination. Lancet. 2021;397:E11–E11.
Schultz NH, Sorvoll IH, Michelsen AE, Munthe LA, Lund-Johansen F, Ahlen MT, et al. Thrombosis and thrombocytopenia after ChAdOx1 nCoV-19 vaccination. New Engl J Med. 2021;384:2124–2130.
See I, Su JR, Lale A, Woo EJ, Guh AY, Shimabukuro TT, et al. US Case reports of cerebral venous sinus thrombosis with thrombocytopenia after Ad26.COV2.S vaccination, March 2 to April 21, 2021. Jama-J Am Med Assoc. 2021;325:2448–56.
Geiben-Lynn R, Greenland JR, Frimpong-Boateng K, Letvin NL. Kinetics of recombinant adenovirus type 5, vaccinia virus, modified vaccinia ankara virus, and DNA antigen expression in vivo and the induction of memory T-lymphocyte responses. Clin Vaccine Immunol. 2008;15:691–6.
Perez P, Marin MQ, Lazaro-Frias A, Sorzano COS, Gomez CE, Esteban M et al. Deletion of vaccinia virus A40R gene improves the immunogenicity of the HIV-1 vaccine candidate MVA-B. Vaccines. 2020;8:70.
Hanke T, McMichael AJ, Samuel RV, Powell LA, McLoughlin L, Crome SJ, et al. Lack of toxicity and persistence in the mouse associated with administration of candidate DNA- and modified vaccinia virus Ankara (MVA)-based HIV vaccines for Kenya. Vaccine. 2002;21:108–14.
Mwau M, Cebere I, Sutton J, Chikoti P, Winstone N, Wee EG, et al. A human immunodeficiency virus 1 (HIV-1) clade A vaccine in clinical trials: stimulation of HIV-specific T-cell responses by DNA and recombinant modified vaccinia virus Ankara (MVA) vaccines in humans. J Gen Virol. 2004;85:911–919.
Ni SH, Bernt K, Gaggar A, Li ZY, Kiem HP, Lieber A. Evaluation of biodistribution and safety of adenovirus vectors containing group B fibers after intravenous injection into baboons. Hum Gene Ther. 2005;16:664–677.
Stone D, Liu Y, Li ZY, Tuve S, Strauss R, Lieber A. Comparison of adenoviruses from species B, C, E, and F after intravenous delivery. Mol Ther. 2007;15:2146–2153.
Lemiale F, Kong WP, Akyurek LM, Ling X, Huang Y, Chakrabarti BK, et al. Enhanced mucosal immunoglobulin A response of intranasal adenoviral vector human immunodeficiency virus vaccine and localization in the central nervous system. J Virol. 2003;77:10078–87.
Shephard E, Burgers WA, Van Harmelen JH, Monroe JE, Greenhalgh T, Williamson C, et al. A multigene HIV type 1 subtype C modified vaccinia Ankara (MVA) vaccine efficiently boosts immune responses to a DNA vaccine in mice. AIDS Res Hum Retroviruses. 2008;24:207–17.
Lovatt A. Applications of quantitative PCR in the biosafety and genetic stability assessment of biotechnology products. J Biotechnol. 2002;82:279–300.
Morata P, Queipo-Ortuno MI, de Dios, Colmenero J. Strategy for optimizing DNA amplification in a peripheral blood PCR assay used for diagnosis of human brucellosis. J Clin Microbiol. 1998;36:2443–6.
Zolla-Pazner S, Gilbert PB. Revisiting the correlate of reduced HIV infection risk in the Rv144 vaccine trial. J Virol. 2019;93:e00629-19.
Someya K, Ami Y, Nakasone T, Izumi Y, Matsuo K, Horibata S, et al. Induction of positive cellular and humoral immune responses by a prime-boost vaccine encoded with simian immunodeficiency virus gag/pol. J Immunol. 2006;176:1784–95.
Collman R, Balliet JW, Gregory SA, Friedman H, Kolson DL, Nathanson N, et al. An infectious molecular clone of an unusual macrophage-tropic and highly cytopathic strain of human immunodeficiency virus type 1. J Virol. 1992;66:7517–21.
Shoji M, Yoshizaki S, Mizuguchi H, Okuda K, Shimada M. Immunogenic comparison of chimeric adenovirus 5/35 vector carrying optimized human immunodeficiency virus clade C genes and various promoters. PLoS One. 2012;7:e30302.
Someya K, Xin KQ, Matsuo K, Okuda K, Yamamoto N, Honda M. A consecutive priming-boosting vaccination of mice with simian immunodeficiency virus (SIV) gag/pol DNA and recombinant vaccinia virus strain DIs elicits effective anti-SIV immunity. J Virol. 2004;78:9842–53.
Perlman J, Gibson C, Pounds SB, Gu Z, Bankowski MJ, Hayden RT. Quantitative real-time PCR detection of adenovirus in clinical blood specimens: a comparison of plasma, whole blood and peripheral blood mononuclear cells. J Clin Virol. 2007;40:295–300.
Saulnier P, Vidaud M, Gautier E, Motte N, Bellet D, Escudier B, et al. Development and validation of a real-time PCR assay for the detection and quantitation of p53 recombinant adenovirus in clinical samples from patients treated with Ad5CMV-p53 (INGN 201). J Virol Methods. 2003;114:55–64.
Xu XX, Shui X, Chen ZH, Shan CQ, Hou YN, Cheng YG. Development and application of a real-time PCR method for pharmacokinetic and biodistribution studies of recombinant adenovirus. Mol Biotechnol. 2009;43:130–7.
Bhaumik S, Gambhir SS. Optical imaging of Renilla luciferase reporter gene expression in living mice. Proc Natl Acad Sci USA. 2002;99:377–82.
Xin KQ, Sekimoto Y, Takahashi T, Mizuguchi H, Ichino M, Yoshida A, et al. Chimeric adenovirus 5/35 vector containing the clade C HIV gag gene induces a cross-reactive immune response against HIV. Vaccine. 2007;25:3809–15.
Acknowledgements
We thank Xian-Xing Xu and Yuan-Guo Cheng for their experimental assistance and Ms. Yuka Takeuchi for her secretary assistance. This work was partially supported by a grant-in-aid from the Ministry of Education, Culture, Sports, Science and Technology of Japan (No. 20K09603). The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript. No additional external funding was received for this study.
Author information
Authors and Affiliations
Contributions
MS, HW, and TU contributed to perform experiments; MS and MI contributed to write the manuscript; MS, NM, and KO contributed to design experiments and discuss the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Shimada, M., Wang, H., Ichino, M. et al. Biodistribution and immunity of adenovirus 5/35 and modified vaccinia Ankara vector vaccines against human immunodeficiency virus 1 clade C. Gene Ther 29, 636–642 (2022). https://doi.org/10.1038/s41434-021-00308-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41434-021-00308-z
