
Abstract
Solitary fibrous tumour (SFT) is an uncommon spindle cell tumour of mesenchymal origin characterised by NAB2-STAT6 gene fusion. Although it was first described in the pleura, it can occur in connective tissue in any part of the body, but rarely presents in the orbit and ocular adnexa. SFT, which is part of the same disease spectrum as other fibroblastic tumours such as giant cell angiofibroma, haemangiopericytoma and fibrous histiocytoma, usually presents as a painless, slow-growing mass in any age group and generally follows a benign course, with a good prognosis after complete excision. However, malignant forms rarely occur. Even for benign tumours a more aggressive clinical behaviour is possible, with relentless infiltrative local growth, frequent recurrence following surgery, and malignant transformation with the potential for metastatic spread. Careful long-term follow-up is essential. The published literature on SFTs of the orbit and ocular adnexa is reviewed, and the aetiology, clinical presentation, epidemiology, radiological features, histopathology, immunohistochemistry, risk stratification, clinical management, and prognosis are discussed, reflecting on our own experience.
摘要
孤立性纤维性瘤(SFT)是一种以NAB2-STAT6基因融合为特征的罕见间充质梭形细胞肿瘤。虽然SFT最早报道出现于胸膜, 但它可发生于身体任何部位的结缔组织中, 但在眼眶和眼附属器中少见。SFT与其他成纤维细胞性肿瘤 (如巨细胞血管纤维瘤、血管外皮细胞瘤和纤维组织细胞瘤) 属于同一疾病谱, 在所有年龄组中通常表现为无痛的、生长缓慢的肿块, 并且通常作为良性疾病治疗, 完全切除后预后良好。然而, 也会发现恶性SFT。即使是良性肿瘤, 也可能出现更具侵袭性的临床表现, 包括持续性局部浸润性生长、术后频繁复发以及具有转移扩散风险的恶变。因此, 对SFT患者进行细致的长期随访很有必要。我们对已发表的有关眼眶和眼附属器SFT的文献进行了回顾, 并对SFT的病因、临床表现、流行病学、放射学特征、组织病理学、免疫组化、危险分层、临床治疗和预后进行了讨论, 提出了自己的见解。
Similar content being viewed by others

Introduction
Solitary fibrous tumours (SFTs) are rare mesenchymal tumours of fibroblastic origin, first documented by Klemperer and Rabin as a distinctive localised pleural based tumour in 1931 [1]. Although they most commonly occur in the thorax, particularly the pleura and mediastinum, they may present almost anywhere in the body [2,3,4]. Orbital involvement is fairly rare and was first described in 1994 [5, 6]. Other documented ophthalmic sites include the conjunctiva [7,8,9,10], caruncle [11], the lacrimal sac [12,13,14,15,16,17,18] and eyelids [9, 19]. Until relatively recently, orbital tumours such as haemangiopericytoma, fibrous histiocytoma and giant cell angiofibroma were thought to be separate entities. However, with recent advances in immunohistochemistry (IHC) and molecular biology, these tumours have now been recognised as variants of a single entity, the SFT [20,21,22]. Although SFTs are generally benign tumours with a good prognosis after complete excision, malignant forms occur [23, 24]. Even in the case of benign tumours a more aggressive clinical behaviour is possible, with relentless infiltrative local growth [25], frequent recurrence following surgery [26,27,28,29,30], malignant transformation [31,32,33,34,35] and the potential for metastatic spread even after many years [27, 36]. Hence, close long-term surveillance is essential. This article aims to provide a comprehensive review of the published literature on SFTs of the orbit and ocular adnexa. The aetiology, clinical presentation, epidemiology, radiological features, histopathology, molecular biology, IHC, risk stratification, clinical management, and prognosis are discussed, with illustrations from a representative case from our institution of a patient with an orbital SFT arising in the superior rectus. Although the superior rectus location is atypical, this case clearly illustrates the common clinical, radiological and pathological features of an orbital SFT.
Search methodology
The published literature on orbital SFTs was evaluated by searching PubMed for papers published in English from 1990 up to 31st January 2022, using the keywords ‘solitary fibrous tumour’, ‘orbit’, ‘lacrimal sac’, ‘conjunctiva’ and ‘eyelid’. Interventional case reports, retrospective case series, clinicopathological studies, gene sequencing studies and authoritative reviews are all included.
Aetiology
SFTs arise de novo with no apparent causative factor [20]. Very little was known about the molecular genetics of these tumours until just over a decade ago, when whole-exome and transcriptome sequencing identified fusion of NGFI-A binding protein 2 (NAB2) and signal transducer and activator of transcription 6 (STAT6) genes due to genomic inversion at the 12q13 gene locus in the vast majority of SFTs [37, 38]. The resultant fusion protein, STAT6, is believed to act as a transcriptional activator through early growth response-mediated pathways [39] and nuclear STAT6 overexpression is a very sensitive and specific biomarker for SFTs [39,40,41]. There have been rare, isolated case reports of orbital SFTs showing accelerated growth in pregnancy, possibly related to a high concentration of progesterone receptors within the tumours [42, 43].
Epidemiology
Head and neck SFTs account for about 6% of all SFTs and, in their multi-institutional retrospective case series comprising 88 SFTs of the head and neck, Smith et al found orbital SFTs made up 25% of the cohort, behind sinonasal tumours which accounted for 30% [44]. Therefore, orbital SFTs account for ~1.5% of SFTs at all sites. Thompson et al. estimate the incidence of orbital SFTs to be 0.2 per million patients per year [27]. Although SFTs are still rare, there has been a steady rise in the number of published cases in recent years due to better immunohistochemical methods facilitating more precise diagnosis. This had led Bernardini et al. to question whether orbital SFTs are that rare after all [45]. In a large retrospective case series comprising 1000 orbital tumours from a single institution in Japan, 5% were SFTs [46]. A database search of all SFT cases reported by the histopathology department at our institution between 2014 and 2021 yielded 94 SFT cases, 2 of which involved the ocular adnexa (2.1%). One tumour (our index case) was from the orbit and another involved the eyelid.
Orbital SFTs appear to have no gender predilection [27, 44]. According to Thompson et al., orbital SFTs present in a younger age group (median 42 years) than the 5–7th decades for other anatomic sites, even when compared to the head and neck region in general where Smith et al. reported a median age of 52 [27, 44]. Rare cases of SFTs affecting the orbit and ocular adnexa presenting in childhood and adolescence have been published [12, 33, 47,48,49,50,51]. Hence, SFT should be in the differential diagnosis of an orbital mass in any age group. 95.6% of all SFTs in the head and neck are ≤5 cm, making them smaller than tumours at other sites [44]. This is largely due to the anatomic confines of the area, especially in the orbit where the tumours will be detected earlier due to signs and symptoms of mass effect. Although most orbital SFTs arise primarily in the orbit they may invade the orbit from adjacent sites, such as the intracranial cavity [52] and sinonasal region [53], or even metastasise from distant sites [54, 55].
Clinical presentation
SFTs are typically slow-growing tumours which can affect any age group, but often present in middle aged patients. Orbital SFTs usually present with painless unilateral proptosis, swelling and globe displacement in the vast majority of cases [26, 27, 56]. Signs and symptoms related to the tumour are usually present for 6–12 months before diagnosis [26, 27]. Blurred vision, ocular motility restriction, a palpable mass, diplopia and ptosis are less common at initial presentation, and headache and epiphora rarely feature [27]. There is no predilection for any particular orbital region but the clinical signs and symptoms are dictated by the precise location within the orbit, for example lacrimal gland [57,58,59,60,61] which may mimic a pleomorphic adenoma [58]. Primary involvement of the eyelid [9, 19], lacrimal apparatus [12,13,14,15,16,17,18] and conjunctiva [7,8,9,10] have also been reported. There are no reports of intrinsic extraocular muscle involvement in the literature. As orbital SFTs progress there is increased mass effect (Fig. 1), and exposure keratopathy as well as optic atrophy may be late findings. Progression of orbital SFTs could also result in invasion of adjacent sites such as the sinonasal cavity, skull base or intracranial cavity [62, 63].

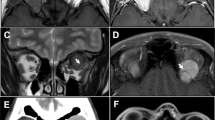
A 41-year-old man with a 1-year history of vertical diplopia, followed by left proptosis and frontal headache for 2–3 months. Note mild inferior displacement of left eye by superior orbital mass. B Progressive tumour growth during 5 years of observation. Note increased left proptosis, worsened hypoglobus, upper lid swelling and significant inferior scleral show. Mild diffuse left conjunctival injection is suggestive of exposure. C Contrast-enhanced coronal MRI scan on presentation revealed a well-circumscribed mass in left superior orbit. The mass is centred on the superior rectus muscle which is not identifiable separate from the mass. There is diffuse contrast enhancement with a few hyperintense streaks. Orbital biopsy confirmed a solitary fibrous tumour. D Contrast-enhanced coronal MRI scan 5 years after presentation shows significant enlargement of the left superior rectus mass, which displays avid heterogenous contrast enhancement and displaces the left optic nerve medially and inferiorly.
Radiology
Radiology plays a vital role in the investigation of patients with orbital SFTs. CT and MRI scans not only help to distinguish orbital SFTs from the myriad of soft tissue tumours encountered in the orbit, but also help with localisation, tumour sizing, planning of surgical intervention and post-operative monitoring.
Orbital SFTs typically appear as unilateral, solitary, well-circumscribed ovoid masses on radiological investigation, displaying avid contrast enhancement on both CT and MRI due to their high vascularity [2, 64, 65]. Both intraconal and extraconal positions are encountered and the tumours may occupy any orbital location. A predilection for the superior extraconal orbit has been reported in several series [26, 64, 66, 67]. However, Alkatan et al. found that the medial orbit was most commonly involved [56].
CT
The key radiological features of orbital SFTs on CT scans are as follows: a well circumscribed ovoid lesion which is either iso-dense or slightly hyper-dense compared to cerebral cortex and displaying avid contrast enhancement which may be homogeneous or heterogeneous (Fig. 2). Kim et al. also reported early washout of contrast material on dual phase CT [2]. Although remodelling of the bony orbit occurs when tumours are large and long-standing, bone destruction only occurs in exceptional cases with aggressive recurrent or malignant tumours [25]. Intralesional calcification is usually absent in SFTs [2].

A Axial CT scan of the orbits 1 year after presentation showing a large well-circumscribed, fusiform mass in the left superior orbit extending to the orbital apex and corresponding to the superior rectus muscle. Note the lesion has similar radiodensity to brain cortex. B Sagittal contrast-enhanced CT scan. Note the avidly enhancing mass in the left superior orbit conforming to the shape of the superior rectus. The mass extends to the muscle origin at the orbital apex and the optic nerve is displaced inferiorly. C Coronal CT scan of orbits 1 year after presentation reveals a large well-circumscribed ovoid mass of brain density in the left superior orbit centred on the superior rectus. The bone is not involved but the left orbit looks expanded. There is no intralesional calcification. D The lesion enhances uniformly with contrast.
MRI
On MRI scanning (Figs. 1 and 3) SFTs generally display homogenous isointense or hypointense signal intensity on T1-weighted images (Fig. 3A) and heterogenous mixed isointense, hypointense or hyperintense signal intensity on T2-weighted images (Fig. 3B and D). Variations in the signal intensity on T2-weighted MR images could reflect the differences in the tissue components, especially collagen (hypointense to isointense signal), and the extent of cystic degeneration (hyperintense signal) within individual tumours [2, 64, 65]. A recent study by Masuno et al. concluded that although no significant correlation was observed between the amount of collagenous tissue and the qualitative evaluation of the signal on T1 and T2-weighted images, kurtosis in the histogram analysis on T2-weighted imaging showed a strong correlation with the amount of collagenous tissue within SFTs [67]. As with CT scanning SFTs display avid contrast enhancement on MRI (Fig. 3C) due to their high vascularity and prominent vascular channels [2, 64, 65]. The time intensity curve (TIC) of dynamic contrast-enhanced MRI (DCE-MRI) shows a slow washout pattern similar to the internal carotid and its large branches, which helps to distinguish orbital SFTs from other orbital tumours. For instance, schwannomas have a persistent or ‘plateau’ TIC pattern on DCE-MRI [64, 66]. In addition, flow voids are sometimes discernible on T2-weighted and contrast enhanced T1-weighted images due to fast flow vessels within the lesions [2, 64]. SFTs display the characteristic of non-restricted diffusion on diffusion-weighted imaging (Fig. 3E) [64, 65].

A Hypointense well-circumscribed ovoid mass in left superior orbit on Coronal T1-weighted MRI. B Coronal T2-weighted MRI reveals intermediate density well-circumscribed lesion in left superior orbit. C Contrast-enhanced and fat-suppressed T1-weighted coronal MRI. The large left superior rectus mass enhances avidly with contrast. Note peripheral ring enhancement (superiorly and medially) and few hyperintense streaks inferiorly. D Axial T2-weighted MRI 5 years after presentation. Note large fusiform mass in left superior orbit. The mass is mostly isointense with brain with some heterogenicity. E Diffusion-weighted MRI scan at presentation showing non-restricted diffusion of the left superior orbital mass.
Ultrasonography
Ultrasound is rarely used in the assessment of orbital SFT. However, 62% of patients in a large series by Blessing et al. underwent ultrasound and the lesions displayed low to medium internal reflectivity [26].
Pathology
Macroscopic appearance
Macroscopically, SFTs are typically firm tumours with a rubbery consistency and, depending on the stromal concentration of collagen and cellularity they may be pale grey, cream, or tan coloured. Figure 4A illustrates the macroscopic appearance of the specimen from our index case following orbital exenteration.

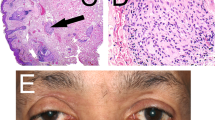
A Macroscopic appearance of longitudinally cut left orbital exenteration specimen. Note the cream-coloured tumour centred on the superior rectus (arrow), infiltrating the orbital fat, and extending to the periorbita superiorly. The tumour extended to the orbital apex posteriorly. Hence the posterior margin was involved, and the orbital apex was debulked piecemeal. B Macroscopic appearance of longitudinally cut left orbital exenteration specimen stained with H + E. Note the mostly basophilic tumour centred on the superior rectus (arrow). C Low power view displaying the typical ‘patternless’ architecture of the solitary fibrous tumour with haphazard growth. H + E (magnification ×200). D High power of bland spindled cells set within a collagenized stroma. Mitotic activity is low. Note a single mitotic figure in this field (arrow). H + E (magnification ×400). E There is myxoid change within the stroma in which many thin-walled open blood vessels could be seen. H + E (magnification ×100). F Orbital solitary fibrous tumour with extraocular muscle fibres evident (arrow). H + E (magnification ×200). G High power showing a very vascular tumour with some blood vessels displaying the classical ‘staghorn’ branching arrangement (arrows). H + E (magnification ×400). H Strong and diffuse cytoplasmic CD34 immunoreactivity (magnification ×200). I Strong and diffuse STAT6 nuclear positivity on immunohistochemistry (magnification ×200).
Histology
Histologically, these tumours of mesenchymal origin usually show patternless, haphazard growth of bland spindle-shaped tumour cells with variable hypo and hypercellular areas and are typically associated with wire-like bundles of collagen, although thick keloid-like collagen may be present (Fig. 4C–F). The ratio of tumour cells to stroma influences the appearance of the tumour. The classic appearance is of a staghorn branching pattern of thin-walled sinusoidal-like vasculature (Fig. 4G) although some dilated vessels have hyalinized thick walls composed of smooth muscle cells [68]. Mitotic figures are variable and may be frequent. Focal necrosis is common [20], cystic degeneration occurs [61, 69,70,71], some SFTs are giant cell-rich [9, 50, 60, 72, 73] and lipomatous differentiation has also been reported [44, 74, 75]. Biphasic tumours comprising both spindle cell and epithelioid architecture have been reported [76, 77], including a case of a recurrent orbital SFT where the epithelioid component was not present when the lesion was first excised [76].
Immunohistochemistry
Although radiology is an extremely useful modality of investigation, it is non-specific. Furthermore, as the lesions are microscopically variable, diagnosing an SFT on histology alone can also be difficult. Therefore, IHC is required for diagnosis.
SFTs usually display diffuse and intense positivity to CD34 (Fig. 4H) due to their mesenchymal origin [5, 21, 26, 27, 56]. However, nuclear STAT6 overexpression is a very sensitive and specific biomarker for SFTs and STAT6 IHC staining (Fig. 4I) is key to separating these tumours from other similar fibroblastic tumours of the orbit [26,27,28, 39,40,41]. Other variably positive markers include vimentin, BCL2 and CD99. SFTs are usually negative for S100 protein, cytokeratins, smooth muscle actin and epithelial membrane antigen [20, 26, 27].
Management
Since orbital SFTs are usually benign, slow-growing tumours conservative management is not unreasonable if the lesion is asymptomatic, not posing a threat to vision and not causing a significant cosmetic blemish.
When intervention is required surgical excision en bloc is the treatment of choice for both primary and recurrent disease to reduce the risk of recurrence, locally aggressive behaviour or malignant transformation which can develop many years later [26, 31, 32, 36, 78, 79]. However, surgery can be challenging because of a friable pseudo-capsule, tenacious attachments and infiltration of orbital fat and important structures in a crowded space, even if the lesion appears well circumscribed radiologically [78]. Furthermore these tumours have a rich vascular supply and have a tendency to bleed during surgery. Therefore, surgery must be weighed up against the risk of blindness, diplopia, and ptosis, especially for persistent or recurrent disease at the orbital apex [26]. The difficulties highlighted above often lead to incomplete excision or piecemeal removal/debulking. In Jackson et al.’s series of seven patients, three tumours were incompletely excised (43%) [78]. Similarly, in Blessing et al.’s series of 21 patients 5 tumours (24%) were removed in piecemeal fashion due to fragmentation during dissection. Interestingly, 3 of the 7 patients with positive surgical margins had persistent or recurrent disease, although 2 of these patients remained asymptomatic and the disease was discovered on routine post-operative imaging [26]. Although en bloc tumour resection is the definitive treatment, residual tumour may remain stable for some time. However, close follow-up is required if complete excision is not achieved [26, 80].
The surgical approach depends on several factors such as tumour size, location, tumour behaviour (soft tissue infiltration) and whether the tumour extends beyond the confines of the orbit. Pre-operative intravascular embolization helps to reduce intraoperative bleeding and facilitate tumour excision [31, 81,82,83,84]. Extirpation of the tumour is often possible without bone removal [85]. However, an osteotomy is sometimes required [86]. Meyer and Riley described the use of a cryoprobe to effect the tumour extraction [87], and an ultrasonic aspirator may also be used [88]. For lacrimal sac tumours extending into the nasolacrimal duct or sinonasal cavity, a combined external and endoscopic approach is desirable [16, 18] and, for tumours involving the skull base or orbital apex a craniofacial approach with neurosurgical involvement is recommended [30]. Orbital exenteration, with or without locoregional flaps, is sometimes required in advanced, aggressive and malignant disease [26, 88, 89].
Adjuvant treatment such as radiotherapy is recommended in high-risk cases. However, its benefit is questionable because there are no available clinical trials, and the value of radiotherapy in the management of orbital SFTs is not yet proven [78]. Stereotactic radiosurgery, which uses precise and calculated doses of radiation to a tightly conformed treatment plan to destroy target lesions without damage to adjacent cells, may have a role to play as adjuvant treatment following incomplete resection or for the treatment of recurrent orbital SFTs, but there is very little published data available [90, 91]. The benefit of adjunctive chemotherapy in the management of orbital SFTs also lacks sufficient evidence and its use must be weighed against the risk of significant side effects. Combination therapy with temozolomide (an oral cytotoxic alkylating agent) and bevacizumab (an anti-VEGF monoclonal antibody) has shown some promise and is generally well tolerated, but myelosuppression is the most common side effect [92].
Risk stratification & prognosis
In general solitary fibrous tumours metastasise in 5–25% of cases [93] but, in a critical review of 47 cases of orbital SFT, Thompson et al estimated the risk of metastatic disease resulting from orbital SFT to be lower at 2.1% [27]. There are few reports in the literature of distant metastases from orbital SFTs. Thompson et al reported 1 case of distant bony metastases to the femur 7 years after excision of an orbital SFT [27]. Tanabe et al. reported a case of an orbital SFT with multiple local recurrences, even after orbital exenteration, followed by radiotherapy and gamma knife treatment, resulting in lung metastases 41 years after primary surgery [36]. Distant metastases have also been localised to the skull base, paravertebral muscles and peritoneum in another case [76]. Although orbital SFTs rarely metastasise, local recurrence is greater in the orbit (26%) than at other sites where the recurrence rate is 10% [27].
Predicting aggressive behaviour of SFT, such as multiple local recurrence, relentless infiltrative growth, malignant transformation and distant metastases, is difficult using clinicopathological features. Several risk stratification models have been devised to predict the risk of aggressive behaviour by SFTs, based on variably employed factors such as mitotic index, tumour size, patient age, cellularity, nuclear pleomorphism, previous radiotherapy and tumour necrosis [27, 93,94,95]. However, these models have been developed mostly in relation to non-orbital SFTs and have been shown to have limited use in predicting the propensity to develop distant metastases for orbital lesions [27, 96]. This is mainly because orbital SFTs are generally smaller (median size 2.6 cm) than their non-orbital counterparts and the patients generally present at a younger age. In proposing a risk stratification model specific for orbital SFTs, Thompson et al. have taken these factors, and the higher risk of local recurrence rather than distant metastases, into account [27]. However, even their model is not perfect and is likely to need further refinement.
Several variants of the NAB2-STAT6 gene fusion which characterises SFTs have been identified, but the clinical implications and prognostic value of these fusion variants is still being explored [97,98,99]. In a recent study Georgiesh et al. found 12 fusion variants that could be divided into two groups based on the STAT6 domain composition in the chimeric protein. STAT6-TAD variants only contained the transactivation domain, and in the STAT6-FULL group most of the STAT6 domains were intact. STAT6-TAD tumours had a higher mitotic count and were associated with an increased risk of recurrence [97]. Another evaluation of NAB2-STAT6 fusion found STAT6-FULL variants were preferentially expressed in older patients, larger tumours and pleuro-pulmonary location, while STAT6-TAD variants were mainly found in younger patients, smaller tumours and meningeal location. However, the authors did not observe preferential expression of any fusion variant in head and neck tumours [98]. Furthermore, that study found no correlation between gene fusion variants and mitotic count or necrosis, and gene fusion variants did not correlate with final patient outcome, nor did they predict adverse clinical events. Park et al. also found no association between NAB2-STAT6 fusion variant and malignant behaviour of SFTs [99]. Therefore, the prognostic value of fusion variants is unclear and needs further exploration but may help to refine the currently available risk stratification models.
The prognostic potential of specific molecular biomarkers in SFTs has also been explored in recent years. The dysregulation of telomerase reverse transcriptase (TERT) through TERT promoter mutations is implicated in the causation of a range of cancers. In one large series TERT promoter mutations were found in 29% of SFTs [100]. Their presence is associated with larger tumour size, necrosis and older patients, as well as being most common in high-risk tumours, but they do not reliably predict clinical outcome [99,100,101]. Inactivation of apoptosis protease activating factor 1, mutation of the tumour suppressor gene P53, and overexpression of BCL6 co-repressor are other examples of biomarkers which may contribute to malignant transformation and play an ancillary role in risk stratification and prognostication [98, 99].
Conclusions
Although there has been a steady rise in the number of published cases in recent years due to better immunohistochemical methods facilitating more precise diagnosis, orbital SFT is still a rare tumour. They present in any orbital location in any age group and should be in the differential diagnosis of any orbital mass. Nuclear STAT6 overexpression is a very sensitive and specific biomarker for SFTs and STAT6 IHC staining is key to separating these tumours from other similar fibroblastic tumours of the orbit [26,27,28, 39,40,41]. Orbital SFTs are generally slow-growing and benign but can display aggressive behaviour with relentless infiltrative local growth [25], frequent recurrence following surgery [26,27,28,29,30], malignant transformation [31,32,33,34,35] and the potential for metastatic spread even after many years [27, 36, 76]. Hence, they may present a significant management challenge. Long-term follow-up is essential because both recurrence and metastatic spread can occur after long disease-free intervals. Predicting the clinical behaviour of these tumours based on their histological appearance alone is difficult and the attempts at risk stratification to date are not fool-proof. The risk stratification models will need further refinement, especially with respect to orbital tumours which are generally smaller, present at an earlier age, have a lower metastatic potential but a greater tendency to local recurrence than non-orbital SFTs [27]. Surgery remains the main stay of treatment and the evidence supporting the use of radiotherapy or chemotherapy as an adjunct to surgery is lacking. Optimal treatment of advanced and metastatic disease is also unclear but should improve with greater research and better understanding of the molecular biology of these tumours.
References
Klemperer P, Rabin CB. Primary neoplasm of the pleura: a report of five cases. Arch Pathol. 1931;11:385–412.
Kim HJ, Kim H-J, Kim Y-D, Yim YJ, Kim ST, Jeon P, et al. Solitary Fibrous Tumor of the Orbit: CT and MR Imaging Findings. Am J Neuroradiol. 2008;29:857–62.
Chick JFB, Chauhan NR, Madan R. Solitary fibrous tumors of the thorax: nomenclature, epidemiology, radiologic and pathologic findings, differential diagnoses, and management. Am J Roentgenol. 2013;200:W238–48.
Gold JS, Antonescu CR, Hajdu C, Ferrone CR, Hussain M, Lewis JJ, et al. Clinicopathologic correlates of solitary fibrous tumors. Cancer 2002;94:1056–68.
Westra WH, Gerald WL, Rosai J. Solitary Fibrous Tumor Consistent CD34 Immunoreactivity and Occurrence in the Orbit. Am J Surg Pathol. 1994;18:992–8.
Dorfman DM, To K, Dickersin GR, Rosenberg AE, Pilch BZ. Solitary Fibrous Tumor of the Orbit. Am J Surg Pathol. 1994;18:281–7.
Knapp AN, Samara WA, Shields CL, Shields JA, Eagle RC. Conjunctival fibrous histiocytoma in an 8-year-old boy. J AAPOS. 2016;20:368–70.
Ali-Ridha A, Brownstein S, O’Connor M, Milman T, Tang T. Benign Solitary Fibrous Histiocytoma of Xanthomatous Subtype of the Perilimbal Conjunctiva and Adjacent Sclera in a Youth. Ocul Oncol Pathol. 2018;4:341–4.
Hayashi N, Borodic G, Karesh JW, Tolentino MJ, Remulla HD, van Wesep RA, et al. Giant cell angiofibroma of the orbit and eyelid. Ophthalmology. 1999;106:1223–9. [cited 2022 Feb 16] Available from https://pubmed.ncbi.nlm.nih.gov/10366097/.
Oh JK, Dehghani A, Shinder R. Solitary fibrous tumor of the conjunctiva. Orbit 2020;39:458–9.
Bonaffini SG, Patel S, Zhou J, Carrasco J. Solitary fibrous tumor of the caruncle: a solitary location. Orbit. 2020:1–3. https://doi.org/10.1080/01676830.2020.1831024.
Santamaria JA, Gallagher CF, Mehta A, Davies BW. Fibrous Histiocytoma of the Lacrimal Sac in an 11-Year-Old Male. Ophthalmic Plast Reconstr Surg. 2018;34:e90–1.
Woo KI, Suh Y-L, Kim Y-D. Solitary Fibrous Tumor of the Lacrimal Sac. Ophthalmic Plast Reconstr Surg. 1999;15:450–3.
Kurdi M, Allen L, Wehrli B, Chakrabarti S. Solitary fibrous tumour of the lacrimal sac presenting with recurrent dacryocystitis. Can J Ophthalmol. 2014;49:e108–10.
Vahdani K, Gupta T, Verity DH, Rose GE. Extension of Masses Involving the Lacrimal Sac to Above the Medial Canthal Tendon. Ophthalmic Plast Reconstr Surg. 2021;37:556–9.
Moriyama M, Kodama S, Hirano T, Suzuki M. Endoscopic-modified medial maxillectomy and its limitation for a solitary fibrous tumor of the lacrimal sac and nasolacrimal duct. Auris Nasus Larynx. 2017;44:370–4.
Bothra N, Dharap R, Ali MJ. Masquerades of Acquired Dacryocystocele. Clin Ophthalmol. 2020;14:1855–8.
Morawala A, Bothra N, Dendukuri G, Ali MJ. Solitary Fibrous Tumors of the Lacrimal Drainage System With Variable Orbital and Sinonasal Extensions: Combined External and Endoscopic Surgical Approach. Ophthalmic Plast Reconstr Surg. 2020;36:403–9.
Kakizaki H, Maden A, Türe M, Yilmaz S, Chan WO. Hemangiopericytoma-solitary fibrous tumor of the eyelid. Ophthalmic Plast Reconstr Surg. 2010;26:46–8.
Mudhar HS, Rushing EJ, Rodriguez F, Vemuganti GK. Solitary fibrous tumour/haemangiopericytoma of the optic nerve sheath. In: Grossniklaus HE, Eberhart CG, Kivelä TT, editors. WHO Classification of Tumours of the Eye. 4th ed [Internet]. Blackwell Publishing Ltd; 2018 [cited 2022 Feb 16]. Available from: https://tumourclassification.iarc.who.int/chaptercontent/40/104.
Furusato E, Valenzuela IA, Fanburg-Smith JC, Auerbach A, Furusato B, Cameron JD, et al. Orbital solitary fibrous tumor: encompassing terminology for hemangiopericytoma, giant cell angiofibroma, and fibrous histiocytoma of the orbit: reappraisal of 41 cases. Hum Pathol. 2011;42:120–8.
Goldsmith JD, van de Rijn M, Syed N. Orbital hemangiopericytoma and solitary fibrous tumor: a morphologic continuum. Int J Surg Pathol. 2001;9:295–302.
Girnita L, Sahlin S, Orrego A, Seregard S. Malignant solitary fibrous tumour of the orbit. Acta Ophthalmol. 2009;87:464–7.
Mascarenhas L, Lopes M, Duarte AM, RomãO H, Honavar M, Resende M, et al. Histologically malignant solitary fibrous tumor of the orbit. Neurochirurgie 2006;52:415–8.
Polito E, Tosi G, Toti P, Schürfeld K, Caporossi A. Orbital solitary fibrous tumor with aggressive behavior. Graefes Arch Clin Exp Ophthalmol. 2002;240:570–4.
Blessing NW, Antonio Bermudez-Magner J, Fernandez MP, Rosenberg AE, Dubovy SR, Johnson TE. Solitary Fibrous Tumor of the Orbit: a Case Series with Clinicopathologic Correlation and Evaluation of STAT6 as a Diagnostic Marker. Ophthalmic Plast Reconstr Surg. 2020;36:164–71.
Thompson LDR, Liou SS, Feldman KA. Orbit solitary fibrous tumor: a proposed risk prediction model based on a case series and comprehensive literature review. Head Neck Pathol. 2021;15:138–52. Available from: https://doi.org/10.1007/s12105-020-01184-6.
Petrovic A, Obéric A, Moulin A, Hamedani M. Ocular adnexal (orbital) solitary fibrous tumor: nuclear STAT6 expression and literature review. Graefes Arch Clin Exp Ophthalmol. 2015;253:1609–17.
Künzel J, Hainz M, Ziebart T, Pitz S, Ihler F, Strieth S, et al. Head and neck solitary fibrous tumors: a rare and challenging entity. Eur Arch Otorhinolaryngol. 2016;273:1589–98.
Yang P, Liu H-C, Qiu E, Wang W, Zhang J-L, Jiang L-B, et al. Factors for postoperative recurrence of orbital solitary fibrous tumor: an analysis of long‐term clinical follow‐up results from a Chinese tertiary hospital. BMC Ophthalmol. 2021;21:61.
Wang X, Qian J, Bi Y, Ping B, Zhang R. Malignant transformation of orbital solitary fibrous tumor. Int Ophthalmol. 2013;33:299–303.
Shen J, Li H, Feng S, Cui H. Orbital solitary fibrous tumor: a clinicopathologic study from a Chinese tertiary hospital with a literature review. Cancer Manag Res. 2018;10:1069–78.
Blandamura S, Alaggio R, Bettini G, Guzzardo V, Valentini E, Bedogni A. Four cases of solitary fibrous tumour of the eye and orbit: one with sarcomatous transformation after radiotherapy and one in a 5-year-old child’s eyelid. J Clin Pathol. 2014;67:263–7.
Chakrabartty H, Singhvi S, Purohit D, Mittal R. Malignant solitary fibrous tumour of orbit. Asian J Neurosurg. 2017;12:55.
Akaike K, Kurisaki-Arakawa A, Hara K, Suehara Y, Takagi T, Mitani K, et al. Distinct clinicopathological features of NAB2-STAT6 fusion gene variants in solitary fibrous tumor with emphasis on the acquisition of highly malignant potential. Hum Pathol. 2015;46:347–56.
Tanabe M, Yoshikawa H, Yamada Y, Oda Y, Sonoda KH. A case of primary orbital solitary fibrous tumor with lung metastases 41 years after initial treatment. Orbit. 2021:1–5. https://doi.org/10.1080/01676830.2021.1954665.
Chmielecki J, Crago AM, Rosenberg M, O’Connor R, Walker SR, Ambrogio L, et al. Whole-exome sequencing identifies a recurrent NAB2-STAT6 fusion in solitary fibrous tumors. Nat Genet. 2013;45:131–2.
Robinson DR, Wu Y-M, Kalyana-Sundaram S, Cao X, Lonigro RJ, Sung Y-S, et al. Identification of Recurrent NAB2-STAT6 Gene Fusions in Solitary Fibrous Tumor by Integrative Sequencing. Nat Genet. 2013;45:180–5.
Doyle LA, Vivero M, Fletcher CD, Mertens F, Hornick JL. Nuclear expression of STAT6 distinguishes solitary fibrous tumor from histologic mimics. Mod Pathol. 2014;27:390–5.
Demicco EG, Harms PW, Patel RM, Smith SC, Ingram D, Torres K, et al. Extensive Survey of STAT6 Expression in a Large Series of Mesenchymal Tumors. Am J Clin Pathol. 2015;143:672–82.
Yoshida A, Tsuta K, Ohno M, Yoshida M, Narita Y, Kawai A, et al. STAT6 Immunohistochemistry Is Helpful in the Diagnosis of Solitary Fibrous Tumors. Am J Surg Pathol. 2014;38:552–9.
Hyde RA, Liu Y, Aakalu VK, Setabutr P. Solitary fibrous tumor of the orbit with growth during pregnancy: a case report. Orbit 2019;38:256–8.
Das J, Sharma A, Deka A, Das D. Solitary fibrous tumor of the orbit presenting in pregnancy. Indian J Ophthalmol. 2009;57:238.
Smith SC, Gooding WE, Elkins M, Patel RM, Harms PW, McDaniel AS, et al. Solitary Fibrous Tumors of the Head and Neck. Am J Surg Pathol. 2017;41:1642–56.
Bernardini FP, de Conciliis C, Schneider S, Kersten RC, Kulwin DR. Solitary fibrous tumor of the orbit. Ophthalmology 2003;110:1442–8.
Goto H, Yamakawa N, Komatsu H, Asakage M, Tsubota K, Ueda S-I, et al. Clinico-epidemiological analysis of 1000 cases of orbital tumors. Jpn J Ophthalmol. 1234;65:704–23. https://doi.org/10.1007/s10384-021-00857-1.
Ribeiro SF, Chahud F, Cruz AAv. Orbital hemangiopericytoma/solitary fibrous tumor in childhood. Ophthalmic Plast Reconstr Surg. 2012;28:e58–60.
Lucci LM, Anderson RL, Harrie RP, Mamalis N, Coffin C, Crandall DC. Solitary Fibrous Tumor of the Orbit in a Child. Ophthalmic Plast Reconstr Surg. 2001;17:369–73.
Rose AM, Kabiru J, Rose GE. Orbit The International Journal on Orbital Disorders, Oculoplastic and Lacrimal Surgery A Rare Case of Orbital Haemangiopericytoma Arising in Childhood A Rare Case of Orbital Haemangiopericytoma Arising in Childhood. Orbit. 2013;32:384–6. [cited 2022 Feb 16]. Available from: https://www.tandfonline.com/action/journalInformation?journalCode=iorb20.
Demirci H, Shields CL, Eagle RC, Shields JA. Giant Cell Angiofibroma, A Variant of Solitary Fibrous Tumor, of the Orbit in a 16-Year-Old Girl. Ophthalmic Plast Reconstr Surg. 2009;25:402–4.
Alexandrakis G, Johnson TE. Recurrent orbital solitary fibrous tumor in a 14-year-old girl. Am J Ophthalmol. 2000;130:373–6.
Ahn JY, Shim JY, Yang WI, Kim TS. Meningeal solitary fibrous tumor as an unusual cause of exophthalmos: case report and review of the literature. Neurosurgery 2001;48:1362–6.
Zeitler D, Kanowitz S, Har-El G. Malignant Solitary Fibrous Tumor of the Nasal Cavity. Skull Base. 2007;17:239–46.
Glazer-Hockstein C, Syed NA, Warhol M, Gausas RE. Malignant Solitary Fibrous Tumor Metastatic to the Orbit. Ophthalmic Plast Reconstr Surg. 2004;20:471–3.
Patel MM, Jakobiec FA, Zakka FR, Du R, Annino DJ, Borboli-Gerogiannis S, et al. Intraorbital Metastasis From Solitary Fibrous Tumor. Ophthalmic Plast Reconstr Surg. 2013;29:e76–9.
Alkatan HM, Alsalamah AK, Almizel A, Alshomar KM, Maktabi AM, ElKhamary SM, et al. Orbital solitary fibrous tumors: a multi-centered histopathological and immunohistochemical analysis with radiological description. Ann Saudi Med. 2020;40:227–33.
Scott IU, Tanenbaum M, Rubin D, Lores E. Solitary Fibrous Tumor of the Lacrimal Gland Fossa. Ophthalmology 1996;103:1613–8.
Feijó ED, Nery AC, de S, Caiado FR, Limongi RM. Solitary fbrous tumor of the lacrimal gland mimicking pleomorphic adenoma. Arq Bras Oftalmol. 2017;80:189–91.
Mupas-Uy J, Kitaguchi Y, Takahashi Y, Takahashi E, Kakizaki H. Solitary fibrous tumor in the lacrimal gland fossa: a case report. Case Rep. Ophthalmol. 2016;7:398–403.
Son DH, Yoo SH, Sa H-S, Cho K-J. A solitary fibrous tumor with giant cells in the lacrimal gland: a case study. Korean J Pathol. 2013;47:158–62.
Gheorghisan-Galateanu A-A, Terzea DC, Burcea I, Dusceac R, Capatina C, Poiana C. Cystic appearance - a new feature of solid fibrous tumours in the lacrimal gland: a case report with literature review. Diagn Pathol. 2019;14:63 https://doi.org/10.1186/s13000-019-0845-x.
Young TK, Hardy TG. Solitary Fibrous Tumor of the Orbit With Intracranial Involvement. Ophthalmic Plast Reconstr Surg. 2011;27:e74–6.
Hayashi S, Kurihara H, Hirato J, Sasaki T. Solitary fibrous tumor of the orbit with extraorbital extension: case report. Neurosurgery 2001;49:1241–5.
Yang BT, Wang YZ, Dong JY, Wang XY, Wang ZC, Bt Y, et al. MRI Study of Solitary Fibrous Tumor in the Orbit. Am J Roentgenol. 2012;199:W506–11. Available from www.ajronline.org.
Liu Y, Tao X, Shi H, Li K. MRI findings of solitary fibrous tumours in the head and neck region. Dentomaxillofac Radio. 2014;43:20130415.
Zhang Z, Shi J, Guo J, Yan F, Fu L, Xian J. Value of MR Imaging in Differentiation between Solitary Fibrous Tumor and Schwannoma in the Orbit. Am J Neuroradiol. 2013;34:1067–71.
Masuno R, Yunaiyama D, Shishido-Hara Y, Yoshimaru D, Maruyama C, Araki Y, et al. Magnetic Resonance Imaging of Orbital Solitary Fibrous Tumors: Radiological-Pathological Correlation Analysis. J Belg Soc Radio. 2021;105:1–7.
Mitamura M, Kase S, Suzuki Y, Sakaguchi T, Suimon Y, Dong Y, et al. Solitary fibrous tumor of the orbit: a clinicopathologic study of two cases with review of the literature. Vivo. 2020;34:3649–54.
Alam S, Backiavathy V, Mukherjee B, Subramanian K. A rare case of giant multicystic solitary fibrous tumor of the orbit. Orbit 2018;37:69–72.
Feuerman JM. Cystic Solitary Fibrous Tumor of the Orbit. Arch Ophthalmol. 2010;128:385–7.
Polomsky M, Sines DT, Dutton JJ. Solitary Fibrous Tumor of the Orbit With Multiple Cavities. Ophthalmic Plast Reconstr Surg. 2013;29:e117–9.
Alsaadi KA, Alwohaib M, Pinto K, Ali RH. Giant cell-rich solitary fibrous tumour of the lacrimal gland with prominent angiomatoid cystic changes and an underlying NAB2ex3-STAT6ex18 fusion. BMJ Case Rep. 2022;15:e247141 https://doi.org/10.1136/bcr-2021-247141.
Mulay K, Honavar S. Orbital solitary fibrous tumor with multinucleate giant cells: case report of an unusual finding in an uncommon tumor. Indian J Pathol Microbiol. 2013;56:282–4.
Guillou L, Gebhard S, Coindre J-M. Lipomatous hemangiopericytoma: a fat-containing variant of solitary fibrous tumor? Clinicopathologic, immunohistochemical, and ultrastructural analysis of a series in favor of a unifying concept. Hum Pathol. 2000;31:1108–15.
Pitchamuthu H, Gonzalez P, Kyle P, Roberts F. Fat-forming variant of solitary fibrous tumour of the orbit: the entity previously known as lipomatous haemangiopericytoma. Eye 2009;23:1479–81.
Parrozzani R, Fusetti S, Montesco C, Favero V, Midena E. Biphasic solitary fibrous tumor of the orbit with distant metastases. Int Ophthalmol. 2013;33:701–5.
Warraich I, Dunn DM, Oliver JW. Solitary Fibrous Tumor of the Orbit With Epithelioid Features. Arch Pathol Lab Med. 2006;130:1039–41.
Jackson CH, Hunt BC, Harris GJ. Fate and Management of Incompletely Excised Solitary Fibrous Tumor of the Orbit: A Case Series and Literature Review. Ophthalmic Plast Reconstr Surg. 2021;37:108–17.
Graue GF, Schubert HD, Kazim M. Correlation between Clinical Features, Imaging and Pathologic Findings in Recurrent Solitary Fibrous Tumor of the Orbit. Orbit 2013;32:375–80.
Tam ES, Chen EC, Nijhawan N, Harvey JT, Howarth D, Oestreicher JH. Solitary fibrous tumor of the orbit: a case series. Orbit 2008;27:426–31.
Wallace KM, Alaraj A, Aakalu VK, Aletich V, Setabutr P. Endovascular Preoperative Embolization of Orbital Hemangiopericytoma With n-Butyl Cyanoacrylate Glue. Ophthalmic Plast Reconstr Surg. 2014;30:e97–100.
Hashemi N, Ling JD, Soparkar C, Sami M, Ellezam B, Klucznik R, et al. Transarterial Onyx Embolization of an Orbital Solitary Fibrous Tumor. Ocul Oncol Pathol. 2015;1:98–102.
Vijitha VS, Kapoor AG, Mittal R, Vangara R. Preoperative embolisation of orbital solitary fibrous tumour. BMJ Case Rep. 2020;13:e235576 https://doi.org/10.1136/bcr-2020-235576.
Demura M, Hayashi Y, Sasagawa Y, Mohri M, Takahira M, Nakada M. Intraorbital solitary fibrous tumor requiring preoperative embolization of feeding artery. Asian J Neurosurg. 2019;14:593–7.
Goldberg RA, Rootman DB, Nassiri N, Samimi DB, Shadpour JM. Orbital Tumors Excision without Bony Marginotomy under Local and General Anesthesia. J Ophthalmol. 2014;2014:424852 https://doi.org/10.1155/2014/424852.
Tanaka K, Yano H, Hayashi H, Hirano A. Total resection combined with osteotomy is more effective for orbital solitary fibrous tumor excision: a report of three cases. Int Ophthalmol. 2018;38:345–51.
Meyer D, Riley F. Solitary fibrous tumor of the orbit: a clinicopathologic entity that warrants both a heightened awareness and an atraumatic surgical removal technique. Orbit 2006;25:45–50.
Navarro-Perea C, Calleja-García C, Bengoa-González Á, Garrido M-C, Mencía-Gutiérrez E, Pérez-Trigo S. Orbital Solitary Fibrous Tumor: Four Case Reports—Clinical and Histopathological Features. Case Rep Ophthalmol Med. 2021;2021:5822859.
Tenekeci G, Sari A, Vayisoglu Y, Serin O. Giant Solitary Fibrous Tumor of Orbit. J Craniofac Surg. 2015;26:e390–2.
Tata A, Cohen-Inbar O, Sheehan JP. Treatment of orbital solitary fibrous tumour with gamma knife radiosurgery and systematic review of literature. BMJ Case Rep. 2016;2016:bcr2016217114 https://doi.org/10.1136/bcr-2016-217114.
Jo K-I, Im YS, Kong D-S, Seol HJ, Nam D-H, Kim Y-D, et al. Multisession Gamma Knife surgery for benign orbital tumors. J Neurosurg. 2012;117:102–7. https://doi.org/10.3171/2012.7.GKS12780.
Park MS, Patel SR, Ludwig JA, Trent JC, Conrad CA, Lazar AJ, et al. Activity of temozolomide and bevacizumab in the treatment of locally advanced, recurrent, and metastatic hemangiopericytoma and malignant solitary fibrous tumor. Cancer 2011;117:4939–47.
Demicco EG, Wagner MJ, Maki RG, Gupta V, Iofin I, Lazar AJ, et al. Risk assessment in solitary fibrous tumors: validation and refinement of a risk stratification model. Mod Pathol. 2017;30:1433–42.
Pasquali S, Gronchi A, Strauss D, Bonvalot S, Jeys L, Stacchiotti S, et al. Resectable extra-pleural and extra-meningeal solitary fibrous tumours: A multi-centre prognostic study. Eur J Surg Oncol. 2016;42:1064–70.
Salas S, Resseguier N, Blay JY, le Cesne A, Italiano A, Chevreau C, et al. Prediction of local and metastatic recurrence in solitary fibrous tumor: construction of a risk calculator in a multicenter cohort from the French Sarcoma Group (FSG) database. Ann Oncol. 2017;28:1779–87.
Sagiv O, Bell D, Guo Y, Su S, Wester ST, Jiang K, et al. Pathological Features and Clinical Course in Patients With Recurrent or Malignant Orbital Solitary Fibrous Tumor/Hemangiopericytoma. Ophthalmic Plast Reconstr Surg. 2019;35:148–54.
Georgiesh T, Namløs HM, Sharma N, Lorenz S, Myklebost O, Bjerkehagen B, et al. Clinical and molecular implications of NAB2-STAT6 fusion variants in solitary fibrous tumour. Pathology. 2021;53:713–9. [cited 2022 Feb 18]. Available from: https://doi.org/10.1016/j.pathol.2020.11.010.
Salguero-Aranda C, Martínez-Reguera P, Marcilla D, de Álava E, Díaz-Martín J. Evaluation of NAB2-STAT6 Fusion Variants and Other Molecular Alterations as Prognostic Biomarkers in a Case Series of 83 Solitary Fibrous Tumors. Cancers (Basel). 2021;13:5237 https://doi.org/10.3390/cancers13205237.
Park HK, Yu DB, Sung M, Oh E, Kim M, Song JY, et al. Molecular changes in solitary fibrous tumor progression. J Mol Med (Berl). 2019;97:1413–25.
Demicco EG, Wani K, Ingram D, Wagner M, Maki RG, Rizzo A, et al. TERT promoter mutations in solitary fibrous tumour. Histopathology 2018;73:843–51.
Bahrami A, Lee S, Schaefer IM, Boland JM, Patton KT, Pounds S, et al. TERT promoter mutations and prognosis in solitary fibrous tumor. Mod Pathol. 2016;29:1511–22.
Author information
Authors and Affiliations
Contributions
CR proposed and devised the scope of the paper, conducted literature search, wrote the paper and made edits. PS contributed to the literature search and writing of the paper, and prepared the illustrations. DGO’D contributed to the literature search, critically reviewed the paper, suggested edits and contributed histopathology slides.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
René, C., Scollo, P. & O’Donovan, D. A review of solitary fibrous tumours of the orbit and ocular adnexa. Eye 37, 858–865 (2023). https://doi.org/10.1038/s41433-022-02160-w
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41433-022-02160-w
This article is cited by
-
A giant orbital solitary fibrous tumor treated by surgical excision: a case report and literature review
Diagnostic Pathology (2023)
-
Presentation of orbital solitary fibrous tumours
Eye (2023)
-
Ocular oncology demystified
Eye (2023)
