
Abstract
Objectives
To identify pathogenic variants in a cohort of 23 black South African children with sporadic primary congenital glaucoma (PCG) using an exome-based approach.
Methods
Children with PCG were recruited from two Paediatric Ophthalmology Clinics in Johannesburg, South Africa. Whole exome sequencing was performed on genomic DNA. Of the 23 children, 19 were male and 19 had bilateral PCG. A variant prioritization strategy was employed whereby variants in known PCG genes (CYP1B1, LTBP2 and TEK) were evaluated first, followed by the identification of putative disease-causing variants in other genes related to eye diseases and phenotypes.
Results
Validated pathogenic variants in the CYP1B1 gene (c.1169 G>A; p.Arg390His) and TEK gene (c.922 G>A; p.Gly308Arg) were identified in one child each. No LTBP2 mutations were identified in this cohort. In silico predictions identified potentially damaging rare variants in genes previously associated with eye development phenotypes or glaucoma in a further 12 children.
Conclusions
This study demonstrates the value of whole exome sequencing in identifying disease-causing variants in African children with PCG. It is the first report of a TEK disease-causing variant in an African PCG patient. Potential causative variants detected in PCG candidate genes warrant further investigation.
Similar content being viewed by others

Introduction
Primary congenital glaucoma (PCG) is a heritable ocular disorder characterised by trabecular meshwork dysgenesis. This isolated developmental anomaly causes an elevated intraocular pressure and patients typically present with the clinical triad of epiphora, photophobia and blepharospasm. Ocular enlargement involving the cornea or the entire globe (buphthalmos) occurs in infants due to the elasticity of immature collagen fibres in the cornea and sclera. The corneal enlargement is associated with curvilinear breaks in Descemet’s membrane (Haab’s striae), corneal oedema and opacification. The final feature, the hallmark of all glaucomatous disease, is optic nerve damage. PCG is more common in males and approximately two thirds of cases are bilateral. It most commonly occurs after 1 month and up to 2 years of age.
PCG is the most common form of childhood glaucoma and untreated cases are a major cause of early onset blindness worldwide [1]. PCG occurs in both sporadic and familial patterns and inheritance is usually autosomal recessive in familial cases. PCG is characterised by both phenotypic and genetic variability that complicate genetic enquiry [2,3,4].
Five genetic loci for PCG (GLC3A–E) have been identified to date. The cytochrome P450 gene (CYP1B1; OMIM #231300) is responsible for the linkage signal at the GLC3A locus [5]. Akarsu et al. mapped the GLC3B locus to chromosome 1p36.2-p36.1 in 1996, but the disease-causing gene at this locus remains unidentified to date [6]. Locus GLC3C was mapped to chromosome 14q24.3 in 2002 [7]. Subsequent studies localised another linkage signal to 14q24.2–24.3, and closer inspection showed that this locus is immediately adjacent to but does not overlap GLC3C [8, 9], leading to the designation of the fourth PCG locus, GLC3D. The gene responsible for GLC3D has been identified as the latent transforming growth factor beta binding protein 2 genes (LTBP2; OMIM #613086) [10], while the GLC3C gene remains unknown. The GLC3E locus was assigned to the tunica interna endothelial cell kinase (TEK) gene (GLC3E; OMIM #617272) after Souma et al. identified heterozygous loss-of-function mutations in the gene in a PCG cohort of 189 families that do not carry mutations in the known PCG causal genes [11].
The diagnosis of PCG is usually made on the basis of a clinical examination under anaesthesia in an infant with suggestive symptoms. However, mutations in CYP1B1 or LTBP2 as autosomal recessive traits (homozygous and compound heterozygous mutations in either gene) or in TEK as an autosomal dominant trait (heterozygous mutation) with variable expressivity would confirm the diagnosis if clinical features are inconclusive [12]. The prevalence of CYP1B1 or LTBP2 mutations varies considerably across different populations [13,14,15,16,17,18,19]. The probability of identifying pathogenic variants in CYP1B1 increases with the presence of a positive family history, parental consanguinity, and bilateral and severe disease [12]. Lower diagnostic yields are, however, expected in cohorts with lower consanguinity rates and in sporadic cases. In these cohorts, an expanded gene panel or exome/genome-based mutation screening approach may therefore be an appropriate strategy to increase the likelihood of identifying the disease-causing mutation in patients presenting with clinical features of PCG.
Data on the genetic aetiology of PCG in African populations remain limited [20, 21]. We describe a cohort of 23 unrelated black South African children with sporadic PCG. We used an exome-based approach to screen for pathogenic mutations in known PCG genes and other genes with putative disease-causing variants that could possibly explain the clinical phenotypes.
Methods
Patient recruitment
The study was approved by the Human Research Ethics Committee (Medical) of the University of the Witwatersrand, South Africa (protocol number M131125), and followed the tenets of the Declaration of Helsinki. The children were recruited from the Paediatric Ophthalmology Clinics at St John Eye Hospital (the eye department of Chris Hani Baragwanath Hospital) and Charlotte Maxeke Johannesburg Academic Hospital. These two public, academic hospitals together serve the greater Johannesburg metropolitan region. Written informed consent was obtained from all patients (and/or their parents/legal guardian) after the nature and possible consequences of the study were explained to them. The diagnosis of PCG required an increased corneal diameter (greater than 11 mm) associated with corneal oedema or Haab striae and an elevated IOP (greater than 21 mmHg or greater than 16 mmHg if measured under general anaesthesia) and/or a vertical cup-to-disc ratio of greater than 0.4. Patients with other ocular abnormalities (like Axenfeld Reiger syndrome, Peters anomaly, aniridia and congenital cataract) or systemic conditions were not included in the study. Detailed information was obtained from the parents and/or a record review was performed on all the participants recording the following details: self-reported ethnicity (home language), presence of consanguinity, family history, medical history, age at onset and diagnosis, laterality, and treatment. Each participant had undergone a complete ophthalmological examination, including an examination under anaesthesia. The following details from ocular examinations performed under anaesthesia were recorded: corneal diameters, intraocular pressures, corneal clarity, pupillary reactions, and vertical cup-to-disc ratio.
None of the children had previously had genetic studies. Genomic DNA was extracted from whole blood using a modified version of the salting out method [22] or from saliva using Oragene DNA Saliva kits (DNA Genotek) and the accompanying prepIT extraction reagents as per the manufacturer’s instructions.
Whole exome sequencing, assembly and variant calling
Targeted exome capture was performed by preparing sequencing libraries from genomic DNA using the NuGEN Ovation Ultralow DR Multiplex protocol followed by a SureSelectXT Human All Exon V5+UTRs (70MB) target enrichment (Agilent). Captured libraries were sequenced on the Illumina HiSeq 2500 (Illumina Inc.) with 76-bp paired-end reads. Reads were mapped to the human hg19 genome assembly using the Burrows-Wheeler Aligner (BWA) [23] and GATK base quality score recalibration, indel realignment, and duplicate removal was applied according to GATK Best Practises [24]. Variants were called with the GATK Unified Genotyper and annotated with the Ensembl Variant Effect Predictor [25]. Variants with a VQSLOD score below 2 and/or a base coverage below 5x were considered low quality calls and consequently not included in the analysis.
Variant prioritization strategy
We excluded the following: (1) non-coding or synonymous variants; (2) variants with a minor allele frequency of more than 2% in the gnomAD database [26]; (3) variants that were reported as benign or likely benign in ClinVar. Variants were evaluated for possible functional impact using the full range of in silico pathogenicity prediction- and conservation scores annotated by the Ensembl Variant Effect Predictor [25].
We then explored the remaining variants, using a model-free approach, for biological relevance. Putative disease-causing variants and variants of uncertain significance (VUSs) were identified using the American College of Medical Genetics and Genomics and the Association for Molecular Pathology guidelines for variant interpretation [27]. For these variants, frequencies in black South African control populations were computed from two data sources: 100 isiZulu individuals as part of the African Genome Variation Project (AGVP) (n = 100) [28]; and 100 South African south-eastern Bantu-speakers who are part of the AWI-Gen study [29] [personal communication MR]. Frequencies were extracted and merged using vcftools [30]. If at least one putative pathogenic allele was observed among the 200 individuals, then that variant is considered less likely to be a PCG causing variant.
Lastly, we analysed all samples with Moon (http://www.diploid.com/moon) to look for variants plausibly linked to individual patients’ clinical phenotypes that were not otherwise identified. Moon is an automated genome interpretation platform that uses artificial intelligence to automatically filter and rank possible pathogenic variants that are associated with patient phenotypes.
Sanger sequencing validations
Validation for the two variants, rs752184169 (TEK) and rs56010818 (CYP1B1) was performed using Sanger sequencing, with primer pairs, 5’-AGTTGGCATGATAGGAGCTCA-3’ and 5’-TGCTGTGCTTTAGGATTTAGGA-3’, and 5’-CCCTGAAATCGCACTGGTG-3’ and 5’-GCTCACTTGCTTTTCTCTCTCC-3’, respectively. The amplicons were sequenced using the BrilliantDye™ Terminator Cycle Sequencing Kit V3.1, BRD3–100/1000 (Nimagen) on the ABI 3500XL Genetic Analyser (Applied Biosystems), according to the manufacturer’s instructions. The AB1 files were analysed using Geneious Prime 2020.1.1 (https://www.geneious.com).
Results
Clinical evaluation
Twenty-three unrelated black South African children were enrolled in the study. Nineteen (83%) were male. Only one child came from a consanguineous union. Another child had a possible (unverified) family history of an affected cousin. The age at diagnosis ranged from a diagnosis at birth to 8 years of age with a median of 6 months of age. Only two of the children were over the age of 3 years at the time of diagnosis. Both had unilateral diseases that had been identified by the parents much earlier but no treatment had been sought. In total, four (17%) of the children presented with unilateral PCG, the remainder had bilateral disease. The corneal diameter of affected eyes ranged from 11.5 mm to 16 mm with a median of 13 mm. The intraocular pressure at the first examination under anaesthesia in affected eyes ranged from 13 mmHg to 41 mmHg with a median of 26.5 mmHg. The cornea was oedematous in all but two of the patients, however, the clear corneas demonstrated Haab’s striae. Four of the patients had an afferent pupillary defect in one eye at the time of presentation. The vertical cup to disc ratio ranged from 0.1 to 1.0 in affected eyes with a median of 0.9. The majority of patients were managed surgically, with trabeculotomy being the most commonly performed procedure. Two patients required multiple surgical procedures including glaucoma drainage devices and cyclophotocoagulation to control the intraocular pressure.
Whole exome sequencing coverage and variant filtering
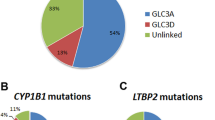
We performed whole exome sequencing on all 23 individuals to an average depth of 43x. On average, 98% and 95% of bases were covered to 5x and 10x within the targeted regions, respectively. The three known PCG genes (CYP1B1, LTBP2 and TEK) were covered adequately in all samples. We identified two variants in known PCG genes (Fig. 1). We identified a pathogenic homozygous c.1169 G>A (p.Arg390His) CYP1B1 variant in patient P009 that had been reported in PCG patients before (rs56010818; ClinVar ID 592512). Patient P017 had a heterozygous c.922 G>A (p.Gly308Arg) variant in TEK that is classified as likely pathogenic. Neither variant was identified in the AWI-Gen or AGVP South African control datasets.

*The sequence is in the reverse direction, therefore, the complementary bases are observed.
We identified VUSs in 12 patients. These were rare variants with damaging in silico predictions in genes that have been associated with eye development phenotypes or glaucoma in the literature without a definitive link to PCG pathogenesis (Table 1). Eight of the variants were identified in at least one individual of either or both of the AWI-Gen or AGVP control datasets suggesting that they are less likely to be pathogenic. The remaining variants were not observed in the South African control datasets, and when present in gnomAD, had low frequencies.
Monoallelic WDR36 and ASB10 variants have been linked to POAG [31, 32]. Patient P004 had rare variants in both WDR36 and ASB10. His diagnosis of PCG was made at 3 months of age. At diagnosis, he had oedematous corneas with diameters of 16 mm in both eyes and had fully cupped discs. His right eye was treated with a trabeculotomy, but he required multiple surgical procedures to control the intraocular pressure in the left eye.
HSPG2 (encoding perlecan; patient P022) and FBN2 (encoding fibrillin-2; patient P023) are microfibril-associated genes and linked to the current theory that microfibril deficiencies cause glaucoma [33, 34]. Fibrillin-2 is the dominant fibrillin in microfibrils of the developing lens [35], while mice deficient in Fbn2 were found to have eye development anomalies, particularly anterior segment dysgenesis [36].
BMP4 (patient P012) is a strong candidate to contribute to anterior segment dysgenesis and other developmental conditions associated with glaucoma as heterozygous deficiency of BMP4 results in anterior segment dysgenesis and elevated IOP in a mouse model [37, 38].
SLC16A12, with a variant identified in patient P027, a 3-year old boy diagnosed with bilateral advanced PCG, is a gene that has previously been associated with anterior segment dysgenesis but it is mostly associated with cataract phenotypes and its association with glaucoma is unclear [39, 40].
VUSs in PIEZO1 were identified in a further two affected boys, patients P018 and P026. PIEZO1 is thought to play a role in optic nerve head astrocyte mechanotransduction [41], and it is hypothesised that this gene might be involved in IOP-mediated glaucomatous neurodegeneration [42, 43].
A 3-month old boy (patient P016) had two heterozygous VUSs in the GALNS gene, neither of which was observed in the Southern African datasets. The GALNS gene (OMIM #253000) is associated with autosomal recessive mucopolysaccharidosis type IVA, also known as Morquio syndrome A. Glaucoma is known to occur in Morquio syndrome, although it is considered rare [44]. The patient in question has, unfortunately, been lost to follow-up and we were unable to perform retrospective phenotyping on him to explore a diagnosis of Morquio syndrome.
The VUSs identified were not validated with Sanger sequencing.
Discussion
In this genetic enquiry into PCG in a South African cohort of unrelated patients, we were able to identify causative mutations in only two of the twenty-three children enrolled in the study. This is, to our knowledge, the first study of its kind in sub-Saharan Africa. Similar studies from around the world suggest that the yield of pathogenic variants in unrelated, sporadic PCG is quite low, with the exception of populations with high levels of consanguinity. In most populations, there is a 10 to 40% chance of identifying a causative mutation [39]. Identifying genes involved in PCG is complicated by incomplete penetrance and variable expressivity of the genetic variants as well as genetic interactions. Since our cohort is relatively small, the detection of 2/23 cases with mutations is not unexpected, and the list of potentially damaging variants may increase the yield if they are validated.
CYP1B1 mutations are the most commonly described in patients with PCG. CYP1B1 is the largest known enzyme of the cytochrome P450 pathway enzymes. This enzyme family is responsible for the metabolism of drugs and dietary compounds and the synthesis of steroid hormones. CYP1B1 is highly expressed in human and murine ocular tissues during development. Cyp1b1 deficient mice exhibit ocular drainage structure abnormalities, which include trabecular meshwork defects and a small or absent Schlemm’s canal, features similar to those reported in human PCG patients [45]. However, the underlying molecular mechanism of CYP1B1’s contribution to the development of the anterior chamber of the eye remains largely unknown [46]. The proportion of patients with CYP1B1 mutations among PCG patients varies in different populations. In an American cohort 15% had pathogenic CYP1B1 mutations [13], in an Australian study 22% [47] and in a European study 35% [48], whereas in a Saudi Arabian study 92% had CYP1B1 mutations [49]. A similar cohort to ours (also from Gauteng province in South Africa) of eleven African children with PCG was investigated for CYP1B1 mutations in 2003 [50] and no mutations were identified.
In this South African study, one infant was identified as being homozygous for a damaging mutation (c.1169 G>A (p.Arg390His)) in the CYP1B1 gene. The mutation is in an important structural core region of the protein and has been classified as a null mutation [51]. This mutation has been reported in PCG patients in India, Pakistan and France [52,53,54], and one patient was a compound heterozygote with a deletion in CYP1B1 as the second mutation [54]. Arg390His has also been reported in the heterozygous state in a 26-year old diagnosed with juvenile-onset primary open-angle glaucoma in France [55]. In our study, the affected infant with this homozygous CYP1B1 mutation (a boy) was born in Johannesburg to first cousins (mother’s father and father’s mother are siblings). The child was the only one in our cohort from a consanguineous union. PCG in this infant was severe and bilateral and was diagnosed before he reached 2 months of age. He was treated with bilateral trabeculotomies. CYP1B1 mutations are generally associated with more severe PCG and with earlier onset. Lopez-Garrido et al., in a large European cohort of PCG patients, found that those with CYP1B1 mutations were younger (most diagnosed at birth) and required more surgical procedures to control their disease [48]. They further demonstrated that recessive CYP1B1 POAG with absent CYP1B1 activity had a worse prognosis.
LTBP2 mutations were first identified in Pakistani and Roma families [10]. Some of these families were found to have the same mutation and also to share a haplotype suggesting a distant common ancestor [10]. LTBP2 is the largest member of the family of latent transforming growth factor (TGF)-beta binding proteins. It has been located to the extracellular matrix, where it performs a regulatory role during elastic fibre assembly [9] and a structural role as an essential component of fibrillin-rich microfibrils in connective tissues [56]. The involvement of LOXL1 in exfoliation glaucoma susceptibility [57] and the recent identification of a mutation in another microfibril-associated gene, ADAMTS10, in a dog model of primary glaucoma [58] led researchers to speculate that defective microfibrils may be an underlying cause of glaucoma [33]. Microfibril-associated genes are therefore plausible candidate glaucoma genes. LTBP2 mutations account for nearly 40% of PCG cases in the Roma ethnic group. In that population, there is a common founder mutation, p.R299X [59]. LTBP2 mutations have also been identified in Indian studies but seem to be uncommon in other populations. None were identified in a Han Chinese study [60] nor in a Turkish and British study [61], nor did we find any in our cohort.
Heterozygous TEK mutations have recently been described in association with PCG [11]. TEK is an angiopoietin receptor that is highly expressed in the Schlemm’s canal endothelium and regulates its development through its interaction with angiopoietin 1 and 2. TEK appears to be essential for the development of the canal of Schlemm. Tek knockout mice do not develop Schlemm’s canal but develop rapidly progressive ocular hypertension, buphthalmos and severe glaucoma. Mice heterozygous for the Tek knockout mutation develop abnormalities and symptoms similar to PCG [11]. The parents of infants with TEK mutations are typically asymptomatic. It is not certain whether this is the result of variable expressivity (some mutation carriers develop glaucoma later on), or reduced penetrance or whether the TEK variants co-occur with heterozygous variants in other glaucoma genes. Kabra et al. subsequently identified heterozygous TEK mutations in 8% of unrelated PCG cases without homozygous mutations in CYP1B1, LTBP2, FOXC1 or MYOC [62]. Some of these TEK mutations, however, co-occurred with heterozygous CYP1B1 mutations. Those cases had a poor visual prognosis, but their parents (with either TEK or CYP1B1 heterozygous mutations) were asymptomatic. In most respects, the presentation of individuals with TEK mutations is usually that of typical PCG with presentation before 3 years, elevated intraocular pressure and enlarged corneal diameters. However, approximately 50% of PCG patients with TEK mutations have unilateral disease. This is in keeping with mutations that demonstrate variable expressivity. Our study identified one child (a boy) with a heterozygous c.922 G>A (p.Gly308Arg) TEK mutation. Multiple in silico tools predict this missense change to be deleterious, and it has only been observed in a heterozygous state in two individuals within the gnomAD dataset [26]. Interestingly, this variant is located 1 bp away from the p.Y307* nonsense variant reported in the Souma et al. study in the EGF-like three domain of the Tek protein [11]. The child in this study was born in Johannesburg to asymptomatic parents who were, unfortunately, not genotyped, so we cannot exclude a de novo mutation. He had bilateral disease. His diagnosis was made at 4 months of age at which stage he had oedematous, enlarged corneas, but minimal optic nerve damage with vertical cup-to-disc ratios of 0.2 in both eyes. He was treated with bilateral trabeculotomies. To our knowledge, this is the first African PCG patient to be reported with a TEK variant.
In eight of the patients in whom we were unable to identify pathogenic mutations in CYP1B1, LTBP2 or TEK, we identified VUSs in genes previously linked to eye disorders, that are rare in gnomAD and were not identified in Southern African control datasets. One of the participants harboured two rare variants in different genes, one of which could potentially be a modifier allele for the other. Another participant had mutations in a gene associated with Morquio Syndrome A. A number of congenital ocular conditions can mimic PCG, therefore, a model-free approach to gene identification using next generation techniques may identify unexpected associations and assist with the diagnosis. It is not clear how, and to what extent, any of the VUSs identified in this study could be linked to glaucoma phenotypes in the absence of validation and segregation information from parental genotypes.
It is evident from population genetics studies that African populations are genetically heterogeneous [63, 64] and, therefore, our findings on the mutation spectrum for PCG among black South Africans is not representative of other African populations.
This study is limited by a small sample size with limited phenotypic information on the parents as well as no parental genotypes. Furthermore, the VUSs identified, while interesting, have not been validated.
In conclusion, this study demonstrates the value of a whole exome sequencing approach to gene and mutation identification in PCG and -related phenotypes. Using this technique, we were able to identify pathogenic mutations in CYP1B1 and TEK in this group of sub-Saharan Africans with PCG. The study also identified VUSs in PCG candidate genes which contribute to the discussion and investigation of mechanisms underlying PCG.
Summary Table
What was known before
-
Primary congenital glaucoma (PCG) is characterised by both phenotypic and genetic variability that complicate genetic enquiry.
-
The yield of pathogenic variants in unrelated, sporadic PCG is quite low, with the exception of populations with high levels of consanguinity.
-
PCG genetics have not been studied in Sub-Saharan Africa.
What this study adds
-
Exome-based mutation screening in this cohort of South African children with PCG successfully identified disease-causing variants in the TEK and CYP1B1 genes.
-
It is the first report of a TEK disease-causing variant in an African PCG patient.
-
Potential causative variants detected in PCG candidate genes warrant further investigation.
References
Gilbert C, Foster A. Childhood blindness in the context of VISION 2020: the right to sight. Bull World Health Organ. 2001;79:227–32.
Ferre-Fernández JJ, Aroca-Aguilar JD, Medina-Trillo C, Bonet-Fernández JM, Méndez-Hernández CD, Morales-Fernández L, et al. Whole-exome sequencing of congenital glaucoma patients reveals hypermorphic variants in GPATCH3, a new gene involved in ocular and craniofacial development. Sci Rep. 2017;7:1–18. https://doi.org/10.1038/srep46175.
Siggs OM, Souzeau E, Pasutto F, Dubowsky A, Smith JE, Taranath D, et al. Prevalence of FOXC1 variants in individuals with a suspected diagnosis of primary congenital glaucoma. JAMA Ophthalmol. 2019;137:348–55.
Cascella R, Strafella C, Germani C, Novelli G, Ricci F, Zampatti S, et al. The genetics and the genomics of primary congenital glaucoma. BioMed Res Int. 2015;2015:321291. https://doi.org/10.1155/2015/321291.
Li N, Zhou Y, Du L, Wei M, Chen X. Overview of Cytochrome P450 1B1 gene mutations in patients with primary congenital glaucoma. Exp Eye Res. 2011;93:572–9.
Akarsu AN, Turacli ME, Aktan SG, Barsoum-Homsy M, Chevrette L, Sayli BS, et al. A second locus (GLC3B) for primary congenital glaucoma (Buphthalmos) maps to the 1p36 region. Hum Mol Genet. 1996;5:1199–203.
Stoilov I, Sarfarazi M. The third genetic locus (GLC3C) for primary congenital glaucoma (PCG) maps to chromosome 14q24. 3. Invest Ophthalmol Vis Sci. 2002;43:3015.
Firasat S, Riazuddin SA, Hejtmancik JF, Riazuddin S. Primary congenital glaucoma localizes to chromosome 14q24. 2-24.3 in two consanguineous Pakistani families. Mol Vis. 2008;14:1659.
Sideek MA, Menz C, Parsi MK, Gibson MA. LTBP-2 competes with tropoelastin for binding to fibulin-5 and heparin, and is a negative modulator of elastinogenesis. Matrix Biol. 2014;34:114–23.
Ali M, McKibbin M, Booth A, Parry DA, Jain P, Riazuddin SA, et al. Null mutations in LTBP2 cause primary congenital glaucoma. Am J Hum Genet. 2009;84:664–71. https://doi.org/10.1016/j.ajhg.2009.03.017.
Souma T, Tompson SW, Thomson BR, Siggs OM, Kizhatil K, Yamaguchi S, et al. Angiopoietin receptor TEK mutations underlie primary congenital glaucoma with variable expressivity. J Clin Invest. 2016;126:2575–87.
Abu-Amero KK & Edward DP. Primary Congenital Glaucoma. 2004. In Adam MP et al. editors. GeneReviews(R) [Internet]. Seattle (WA): University of Washington. 1993–2022.
Lim S-H, Tran-Viet K-N, Yanovitch TL, Freedman SF, Klemm T, Call W, et al. CYP1B1, MYOC, and LTBP2 mutations in primary congenital glaucoma patients in the United States. Am J Ophthalmol. 2013;155:508–17. e505.
Do T, Shei W, Chau PTM, Le Trang D, Yong VH, Ng XY, et al. CYP1B1 and MYOC mutations in vietnamese primary congenital glaucoma patients. J Glaucoma. 2016;25:e491–e498.
Sitorus R, Ardjo S, Lorenz B, Preising M. CYP1B1 gene analysis in primary congenital glaucoma in Indonesian and European patients. J Med Genet. 2003;40:e9–e9.
Chakrabarti S, Kaur K, Kaur I, Mandal AK, Parikh RS, Thomas R, et al. Globally, CYP1B1 mutations in primary congenital glaucoma are strongly structured by geographic and haplotype backgrounds. Invest Ophthalmol Vis Sci. 2006;47:43–47. https://doi.org/10.1167/iovs.05-0912.
Stoilov IR, Costa VP, Vasconcellos JP, Melo MB, Betinjane AJ, Carani JC, et al. Molecular genetics of primary congenital glaucoma in Brazil. Invest Ophthalmol Vis Sci. 2002;43:1820–7.
Plasilova M, Stoilov I, Sarfarazi M, Kadasi L, Ferakova E, Ferak V. Identification of a single ancestral CYP1B1 mutation in Slovak Gypsies (Roms) affected with primary congenital glaucoma. J Med Genet. 1999;36:290–4.
Abu-Amero KK, Osman EA, Mousa A, Wheeler J, Whigham B, Allingham RR, et al. Screening of CYP1B1 and LTBP2 genes in Saudi families with primary congenital glaucoma: genotype-phenotype correlation. Mol Vis. 2011;17:2911.
Bouyacoub Y, Ben Yahia S, Abroug N, Kahloun R, Kefi R, Khairallah M, et al. CYP1B1 gene mutations causing primary congenital glaucoma in Tunisia. Ann Hum Genet. 2014;78:255–63.
Hadrami M, Bonnet C, Zeitz C, Veten F, Biya M, Hamed CT, et al. Mutation profile of glaucoma candidate genes in Mauritanian families with primary congenital glaucoma. Mol Vis. 2019;25:373.
Miller S, Dykes D, Polesky H. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988;16:1215.
Li H, Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics. 2009;25:1754–60.
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–303.
McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GR, Thormann A, et al. The ensembl variant effect predictor. Genome Biol. 2016;17:1–14.
Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581:434–43.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–23.
Gurdasani D, Carstensen T, Tekola-Ayele F, Pagani L, Tachmazidou I, Hatzikotoulas K, et al. The African Genome Variation Project shapes medical genetics in Africa. Nature. 2015;517:327–32. https://doi.org/10.1038/nature13997.
Ramsay M, Crowther N, Tambo E, Agongo G, Baloyi V, Dikotope S, et al. H3Africa AWI-Gen Collaborative Centre: a resource to study the interplay between genomic and environmental risk factors for cardiometabolic diseases in four sub-Saharan African countries. Glob Health Epidemiol Genom. 2016;1:e20. https://doi.org/10.1017/gheg.2016.17.
Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, et al. The variant call format and VCFtools. Bioinformatics. 2011;27:2156–8. https://doi.org/10.1093/bioinformatics/btr330.
Monemi S, Spaeth G, DaSilva A, Popinchalk S, Ilitchev E, Liebmann J, et al. Identification of a novel adult-onset primary open-angle glaucoma (POAG) gene on 5q22. 1. Hum Mol Genet. 2005;14:725–33.
Pasutto F, Keller KE, Weisschuh N, Sticht H, Samples JR, Yang Y-F, et al. Variants in ASB10 are associated with open-angle glaucoma. Hum Mol Genet. 2012;21:1336–49.
Kuchtey J, Kuchtey RW. The microfibril hypothesis of glaucoma: implications for treatment of elevated intraocular pressure. J Ocul Pharm Ther. 2014;30:170–80.
Ashworth JL, Kielty CM, McLeod D. Fibrillin and the eye. Br J Ophthalmol. 2000;84:1312–7.
Shi Y, Tu Y, De Maria A, Mecham RP, Bassnett S. Development, composition, and structural arrangements of the ciliary zonule of the mouse. Invest Ophthalmol Vis Sci. 2013;54:2504–15.
Shi Y, Tu Y, Mecham RP, Bassnett S. Ocular phenotype of Fbn2-null mice. Invest Ophthalmol Vis Sci. 2013;54:7163–73.
Chang B, Smith RS, Peters M, Savinova OV, Hawes NL, Zabaleta A, et al. Haploinsufficient Bmp4 ocular phenotypes include anterior segment dysgenesis with elevated intraocular pressure. BMC Genet. 2001;2:1–12.
Wordinger RJ, Fleenor DL, Hellberg PE, Pang I-H, Tovar TO, Zode GS, et al. Effects of TGF-β2, BMP-4, and gremlin in the trabecular meshwork: implications for glaucoma. Invest Ophthalmol Vis Sci. 2007;48:1191–200.
Yu-Wai-Man C, Arno G, Brookes J, Garcia-Feijoo J, Khaw PT, Moosajee M. Primary congenital glaucoma including next-generation sequencing-based approaches: clinical utility gene card. Eur J Hum Genet. 2018;26:1713–8.
Kloeckener-Gruissem B, Vandekerckhove K, Nürnberg G, Neidhardt J, Zeitz C, Nürnberg P, et al. Mutation of solute carrier SLC16A12 associates with a syndrome combining juvenile cataract with microcornea and renal glucosuria. Am J Hum Genet. 2008;82:772–9.
Liu Y, Liu J, Clark AF, Yang Y. Piezo1 plays a role in optic nerve head astrocyte mechanotransduction. Invest Ophthalmol Vis Sci. 2019;60:6185–6185.
Choi HJ, Sun D, Jakobs TC. Astrocytes in the optic nerve head express putative mechanosensitive channels. Mol Vis. 2015;21:749.
Morozumi W, Inagaki S, Iwata Y, Nakamura S, Hara H, Shimazawa M. Piezo channel plays a part in retinal ganglion cell damage. Exp Eye Res. 2020;191:107900.
Tomatsu S, Pitz S, Hampel U. Ophthalmological findings in mucopolysaccharidoses. J Clin Med. 2019;8:1467.
Libby RT, Smith RS, Savinova OV, Zabaleta A, Martin JE, Gonzalez FJ, et al. Modification of ocular defects in mouse developmental glaucoma models by tyrosinase. Science. 2003;299:1578–81.
Faiq MA, Dada R, Qadri R, Dada T. CYP1B1-mediated pathobiology of primary congenital glaucoma. J Curr Glaucoma Pr. 2015;9:77.
Dimasi D, Hewitt A, Straga T, Pater J, MacKinnon J, Elder J, et al. Prevalence of CYP1B1 mutations in Australian patients with primary congenital glaucoma. Clin Genet. 2007;72:255–60.
López-Garrido M-P, Medina-Trillo C, Morales-Fernandez L, Garcia-Feijoo J, Martínez-de-la-Casa J-M, García-Antón M, et al. Null CYP1B1 genotypes in primary congenital and nondominant juvenile glaucoma. Ophthalmology. 2013;120:716–23.
Bejjani BA, Stockton DW, Lewis RA, Tomey KF, Dueker DK, Jabak M, et al. Multiple CYP1B1 mutations and incomplete penetrance in an inbred population segregating primary congenital glaucoma suggest frequent de novo events and a dominant modifier locus. Hum Mol Genet. 2000;9:367–74.
Molecular Genetic Defects in True Primary Congenital Glaucoma and Juvenile Congenital Glaucoma. MMed thesis, Medical University of Southern Africa (Medunsa), (2003).
Medina-Trillo C, Ferre-Fernández JJ, Aroca-Aguilar JD, Bonet-Fernández JM, Escribano J. Functional characterization of eight rare missense CYP1B1 variants involved in congenital glaucoma and their association with null genotypes. Acta Ophthalmol. 2016;94:e555–e560. https://doi.org/10.1111/aos.13017.
Stoilov I, Akarsu AN, Alozie I, Child A, Barsoum-Homsy M, Turacli ME, et al. Sequence analysis and homology modeling suggest that primary congenital glaucoma on 2p21 results from mutations disrupting either the hinge region or the conserved core structures of cytochrome P4501B1. Am J Hum Genet. 1998;62:573–84. https://doi.org/10.1086/301764.
Reddy ABM, Kaur K, Mandal AK, Panicker SG, Thomas R, Hasnain SE, et al. Mutation spectrum of the CYP1B1 gene in Indian primary congenital glaucoma patients. Mol Vis. 2004;10:696–702.
Colomb E, Kaplan J, Garchon HJ. Novel cytochrome P450 1B1 (CYP1B1) mutations in patients with primary congenital glaucoma in France. Hum Mutat. 2003;22:496. https://doi.org/10.1002/humu.9197.
Melki R, Colomb E, Lefort N, Brezin A, Garchon H. CYP1B1 mutations in French patients with early-onset primary open-angle glaucoma. J Med Genet. 2004;41:647–51.
Inoue T, Ohbayashi T, Fujikawa Y, Yoshida H, Akama TO, Noda K, et al. Latent TGF-β binding protein-2 is essential for the development of ciliary zonule microfibrils. Hum Mol Genet. 2014;23:5672–82.
Thorleifsson G, Magnusson KP, Sulem P, Walters GB, Gudbjartsson DF, Stefansson H, et al. Common sequence variants in the LOXL1 gene confer susceptibility to exfoliation glaucoma. Science. 2007;317:1397–400.
Ahonen SJ, Kaukonen M, Nussdorfer FD, Harman CD, Komáromy AM, Lohi H. A novel missense mutation in ADAMTS10 in Norwegian Elkhound primary glaucoma. PLoS One. 2014;9:e111941.
Azmanov DN, Dimitrova S, Florez L, Cherninkova S, Draganov D, Morar B, et al. LTBP2 and CYP1B1 mutations and associated ocular phenotypes in the Roma/Gypsy founder population. Eur J Hum Genet. 2011;19:326–33.
Chen X, Chen Y, Fan BJ, Xia M, Wang L, Sun X. Screening of the LTBP2 gene in 214 Chinese sporadic CYP1B1-negative patients with primary congenital glaucoma. Mol Vis. 2016;22:528.
Sharafieh R, Child AH, Khaw PT, Fleck B, Sarfarazi M. LTBP2 gene analysis in the GLC3C-linked family and 94 CYP1B1-negative cases with primary congenital glaucoma. Ophthalmic Genet. 2013;34:14–20. https://doi.org/10.3109/13816810.2012.716486.
Kabra M, Zhang W, Rathi S, Mandal AK, Senthil S, Pyatla G, et al. Angiopoietin receptor TEK interacts with CYP1B1 in primary congenital glaucoma. Hum Genet. 2017;136:941–9.
Choudhury A, Aron S, Botigué LR, Sengupta D, Botha G, Bensellak T, et al. High-depth African genomes inform human migration and health. Nature. 2020;586:741–8. https://doi.org/10.1038/s41586-020-2859-7.
Choudhury A, Ramsay M, Hazelhurst S, Aron S, Bardien S, Botha G, et al. Whole-genome sequencing for an enhanced understanding of genetic variation among South Africans. Nat Commun. 2017;8:1–12. https://doi.org/10.1038/s41467-017-00663-9.
Acknowledgements
We would like to thank the patients and their parents for their participation and cooperation. MR is a South African Research Chair in Genomics and Bioinformatics of African populations hosted by the University of the Witwatersrand, funded by the Department of Science and Technology and administered by National Research Foundation of South Africa (NRF). SW had a Carnegie Clinician Scientist post-doctoral fellowship. Exome sequencing and analysis was performed during NC’s participation in the Novartis and University of Basel Next Generation Scientist program, a 3-month research internship in Basel, Switzerland that aims to build scientific and leadership capability in emerging country scientists.
Funding
Research reported in this publication was supported by the Fogarty International Centre of the National Institutes of Health under Award Number D43 TW008330 and the Carnegie Corporation. The content is solely the responsibility of the authors and does not necessarily represent the official views of the Fogarty International Centre, The Carnegie Corporation or the National Institutes of Health or the NRF.
Author information
Authors and Affiliations
Contributions
The contributing authors (NC, SG, MH, J-TB, MR and SW) meet all of the following criteria: Conceived and/or designed the work that led to the submission, acquired data, and/or played an important role in interpreting the results. Drafted or revised the manuscript. Approved the final version. Agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Carstens, N., Goolam, S., Hulley, M. et al. Exome-based mutation screening in South African children with primary congenital glaucoma. Eye 37, 362–368 (2023). https://doi.org/10.1038/s41433-022-01941-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41433-022-01941-7
