
Abstract
Background
Down syndrome is a common multigene, multisystem disorder associated with abnormalities of visual function and characteristic changes in the majority of tissues in the eye. Historic descriptions of macular structure in Down syndrome have been variable, but optical coherence tomography allows increasingly detailed characterization of retinal architecture in vivo. We demonstrate the feasibility of retinal imaging in children with Down syndrome using handheld OCT in an outpatient clinical setting, and describe the foveal and parafoveal retinal architecture in this group.
Methods
Fourteen White British children aged between 4 and 11 with Down syndrome were recruited to have handheld SD-OCT retinal imaging performed at a single centre in an outpatient clinical setting. The thickness of the retinal layers at the fovea and parafovea was analysed using segmentation software, and compared with age-matched controls from a previously published normative UK dataset.
Results
Sixty-seven percent of the children studied had grade 1 foveal hypoplasia. At the fovea, the ganglion cell layer (p = 0.002) and inner nuclear layer (p < 0.001) were thickened relative to the control group. At the parafovea, there was thickening of the retina attributable to numerous layers in both the inner and outer retina, which remained significant after Bonferroni correction.
Conclusion
OCT imaging of children with Down syndrome in an outpatient setting is feasible. There is a high incidence of foveal hypoplasia in this group, associated with thickening of the ganglion cell and inner nuclear layers at the fovea.
Similar content being viewed by others


Introduction
Down syndrome, or trisomy 21, is caused by the presence of all or part of the third copy of chromosome 21 [1]. It is the most frequently occurring chromosomal abnormality in humans and affects between 1 in 400 and 1 in 1500 babies born in different populations, depending on maternal age and prenatal screening schedules [2]. In Europe, the incidence of Down syndrome is 1 in 1000 live births, accounting for 8% of all registered cases of congenital anomaly [3].
There are reported associations between Down syndrome and abnormalities in the majority of the tissues of the eye, including distinctive changes to eyelid anatomy, corneal ectasia [4], infantile glaucoma [5], iris stromal hyperplasia (Brushfield spots) [3], juvenile cataracts [6], retinal vascular anomalies [7], optic disc pallor [6], and foveal hypoplasia [8]. Nystagmus is commonly reported in 10–30% of patients with Down syndrome [9,10,11,12,13,14] although it has been suggested that the true prevalence may be significantly higher [6, 15]. Strabismus affects 22–57% of patients with Down syndrome [16,17,18]. Abnormalities of visual function are common in this group, including abnormalities of visual acuity [12], contrast sensitivity [19], vernier acuity [20], and accommodative ability [21, 22].
Development of the fovea begins around 24–28 weeks of gestation, and continues until at least 4 years of age [23]. The development of the macular region may be disrupted by premature birth [24], altered visual input [25], and by myriad genetic conditions [26].
Reports on retinal structure in children with Down syndrome are highly variable. Foveal hypoplasia has been reported in 1.2% of a UK case series [11] and in 9.2% of a Lithuanian case series [6], both based on ophthalmoscopic appearance.
The advent of handheld high resolution spectral-domain ocular coherence tomography (SD-OCT) has the potential to improve our understanding of ocular development and paediatric ophthalmic care, in the same way that table-mounted OCT transformed our understanding of adult retinal structure and pathology [27]. The reliability and reproducibility of handheld OCT to assess the retinal layer in children from birth to 13 years has been demonstrated by numerous groups [27,28,29,30].
The primary aim of this study was to assess the feasibility of imaging the retinal layers with handheld OCT in non-sedated children with Down syndrome. The secondary aim was to compare the retinal microstructure with previously reported normative data for children without Down syndrome in order to identify any differences between the two cohorts.
Methods
Study design and participants
This is a prospective observational study, which recruited children between the ages of 3 and 11 years from the Southampton Down syndrome research registry. The registry included children recruited from community, hospital and parent support group settings to provide a representative sample [31]. The presence or absence of visual impairment was not a focus of the original study. All participants had undergone previous routine vision surveillance and any participant with an additional ocular, a neurologic or metabolic abnormality that could affect ocular development was excluded.
Examination
All participants were examined between 1200 h and 1700 h to limit circadian influences on OCT measures [32, 33]. Participants underwent a full orthoptic and ophthalmologic examination including measurement of visual acuity, measurement of refractive error, and dilated fundus examination. All examinations were performed by the same orthoptist (DO) and ophthalmologist (RN). Visual acuity was assessed in younger children by preferential looking using Teller acuity cards. In cooperative children, single optotypes (single Kay pictures) or crowded optotypes (crowded Kay pictures) were used to measure visual acuity. Mydriasis was achieved with cyclopentolate 1% drops. A hand-held SD-OCT system (Envisu C2300, Leica Microsystems, Wetzlar, Germany), was used to obtain a 10 mm×10 mm volumetric scan (consisting of 100 B-scans and 1000 A-scans per B-scan) of the fovea and optic nerve region in horizontal and vertical planes as previously described [34]. In all cases the OCT scan was obtained from the right eye first, followed by the left eye.
SD-OCT acquisition, segmentation analysis and statistics
The acquired OCT images were exported from the Bioptigen OCT software and imported into ImageJ software (http://imagej.nih.gov/ij/; provided in the public domain by the National Institutes of Health, Bethesda, MD, USA) where retinal layer segmentation was manually performed by two of the authors (RN and DO) using a customised macro. Segmentation data were compared to confirm internal consistency, and were exported to Excel V.16.3 (Microsoft, Redmond, WA, USA) for evaluation, and to SPSS V.16.0 (SPSS, Chicago, Il, USA) for statistical analysis.
Images were deemed of a sufficient quality for analysis if there were adequate adjacent tomograms to determine centration, and both of the segmenting authors agreed that they were able to define the boundaries of the layers within the Bioptigen software.
For each child, the images from the right eye only were analysed, unless the images were deemed of insufficient quality for reliable analysis in which case the images from the left eye were used for statistical analysis.
All retinal layers were segmented at the fovea, and the parafovea 1000 μm nasal and temporal to the fovea, after adjusting for estimated axial length as previously described [27].
For comparison with previously published normative data, weighted averages were calculated for mean layer thicknesses and the standard deviations of the means to account for the age group composition of our cohort. The distribution was assessed with the Shapiro-Wilk test, and differences between the participants and normative data were explored using a two-sample t test. Bonferroni correction was used due to the large number of layers measured.
Ethics
The study adhered to the tenets of the Declaration of Helsinki, and the research was approved by UK Health Research Authority (IRA reference 17/SC/004) and the University of Southampton (ERGO 30768). Parents/legal guardians gave consent on behalf of their child.
Results
Fourteen participants with Down syndrome were recruited (Table 1). The mean age was 6 years 10 months (range 55–130 m, SD 24 m). Fifty-seven percentage were male. All participants were White British.
SD-OCT images of a sufficient quality for analysis were obtained in 12/14 children (86%). The two children with failed imaging were aged 63 months and 82 months; the reason for imaging failure in both cases was that they were distressed on the day of assessment and would not permit adequate imaging for the study.
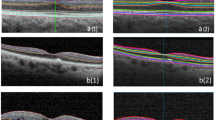
Sixty-seven percentage of eyes examined had foveal hypoplasia. All cases of foveal hypoplasia were bilateral and grade 1 according to a previously described OCT grading system [35]. An example of foveal hypoplasia seen in the cohort is shown in Fig. 1. While some cases of foveal hypoplasia clearly fitted the descriptor of type 1a (including the case shown in Figs. 1) or 1b, most cases were felt to be of an intermediate appearance and so it was judged that reliable subcategorisation was not possible.

Grade 1 foveal hypoplasia is demonstrated, with continuation of the inner retinal layers through the fovea. The retinal layers are labelled as follows: retinal nerve fibre layer (RNFL), ganglion cell layer (GCL), inner plexiform layer (IPL), inner nuclear layer (INL), outer plexiform layer (OPL), outer nuclear layer (ONL), inner photoreceptor segments (IS), outer photoreceptor segments (OS), and retinal pigment epithelium (RPE).
At the fovea, the ganglion cell layer (p = 0.02) and inner nuclear layer (p < 0.001) were significantly thicker than previously reported in children without Down syndrome (Table 2, Fig. 2). The remainder of the outer retinal layers were not significantly different from the normative cohort referenced, after Bonferroni correction.

Bars denote 95% confidence intervals. Significantly different segments after Bonferroni correction highlighted with an asterisk.
At the nasal parafovea (1000 microns nasal to the centre of the fovea), the retinal nerve fibre layer (p < 0.0001), ganglion cell layer (p < 0.0001), outer nuclear layer (p = 0.0004), inner segment (p = 0.0001), outer segment (p < 0.0001) and total retinal thickness (p = 0.0001) were all significantly thicker than in the normative cohort (Table 2, Fig. 3). At the temporal parafovea, the retinal nerve fibre layer (p < 0.0001), ganglion cell layer (p = 0.0001), outer segment (p < 0.0001) and total retinal thickness (p = 0.0002) were all significantly thicker than in the normative cohort.

Bars denote 95% confidence intervals. Significantly different segments after Bonferroni correction highlighted with an asterisk.
Discussion
In this study, we report on the thickness of each of the retinal layers at the fovea in White British children with Down syndrome, as measured by handheld SD-OCT. All data were collected in an outpatient setting which reflects clinical practice. We demonstrate the feasibility of capturing OCT data in young children with Down syndrome in such a setting, and we present evidence that the incidence of foveal hypoplasia in children with Down syndrome is significantly higher than historically reported.
There are few previous reports on retinal microstructure in children with Down syndrome. Three previous studies have reported retinal macular structural features as observed using OCT in children with Down syndrome. Laguna et al. [36] described a thickening of the central macula in three individuals with Down syndrome (mean age 29.2 years ± 16.6 years) compared with age-matched controls, and found a similar thickening of the macula in a murine model of Down syndrome. Histological examination of the murine model showed that the inner retina (and not the outer retina) was hyperplastic due to reduced developmental apoptosis, and the team demonstrated that triplication of the Dyrk1a gene was necessary and sufficient to cause this phenotype. O’Brien et al. [37]. found an increased central subfield macular thickness in 17 children with Down syndrome (mean age 11.0 years ± 3.1 years) compared with age-matched controls. Their data did not enable them to comment on individual retinal layers or foveal contours, but they reported that the increased macular thickness was attributable to both the inner and outer retina. This is in contrast with the findings of Laguna et al. [36]. who found that in a mouse model of Down syndrome only the inner retinal layers contributed significantly to macular thickening. More recently, Mangalesh et al. [38]. described the macular OCT appearance in a cohort of 19 South Asian children with Down syndrome (mean age 24 months ± 3.1 months), finding abnormal foveal architecture and persistence of the inner retinal layers.
Our findings are consistent with these previous reports, and the segmentation performed in this study localises the increased foveal thickness to the ganglion cell and inner nuclear layers.
In normal foveal development, the thickness and number of inner retinal layers at the foveal centre decreases between 31 and 42 weeks postmenstrual age due to inner retinal cell displacement centrifugally, causing deepening of the foveal pit [23, 39, 40]. Apoptosis in the ganglion cell and inner nuclear layers does not appear to be a significant factor in foveal depression morphogenesis during normal development [41], rather it is largely due to migration of inner retinal neurons away from the foveal centre, and later migration of cones towards the pit centre [42].
A characteristic abnormality in human Down syndrome is delayed and disorganised cortical lamination [43, 44]. The mechanisms by which this occurs are multifactorial and are not fully characterised, but similar mechanisms may contribute to the abnormalities seen in foveal lamination. Migration deficits of neurons during corticogenesis have been observed in murine models of Down syndrome [45] and more recently defective migration has been observed in cortical GABA-ergic interneurons derived from induced pluripotent stem cells from people with Down syndrome [46]. In the latter study, correction of PAK1 overexpression reversed the migration deficit in vitro.
We hypothesise that the persistence of the ganglion cell and inner nuclear layers in children with Down syndrome is a multifactorial phenomenon caused by overexpression of multiple genes of chromosome 21. Neuronal migration defects including those linked to the PAK1 pathway may result in the persistence of cell bodies such as GABA-ergic amacrine cells in the inner retinal layers of the fovea in children with Down syndrome. In addition, reduced developmental apoptosis linked to overexpression of genes, including Dyrk1a may contribute to the persistence of the inner nuclear and ganglion cell layers.
Although numerous abnormalities of ocular anatomy and function have been associated with Down syndrome, it is notable that some defects in foveal development, such as fovea plana, may have no measurable effect on visual acuity [47, 48]. However, we note recent work finding that structural grading of foveal hypoplasia based on handheld OCT is a good predictor of future visual acuity in idiopathic infantile nystagmus [49]. Our cohort was not large enough to explore whether the presence of foveal hypoplasia correlated with visual function, strabismus or nystagmus in affected individuals, and further research is needed to answer this question.
Strengths of our study include its ‘real-world’ outpatient clinical setting, and the age group of the children studied. Previous studies [38] have assessed younger age groups, and foveal pit formation may not be complete until age 2 [23].
Limitations of this study include the relatively small cohort size which precluded further analysis of the correlation between macular architecture and visual function, and is also likely to have impacted on our ability to demonstrate statistically significant differences for some retinal layers after employing Bonferroni correction. We recommend caution in generalising results from this small sample. We acknowledge the lack of a prospective control group, relying instead on age-matched controls from published normative UK datasets. While the ethnically homogeneous cohort strengthens the reliability of our findings for the specific population in question, the conclusions may not be equally applicable to other populations.
As Down syndrome is a multigene, multisystem disorder, understanding the high degree of variability across functional neurological phenotypes remains a significant challenge [50]. We suggest that our observation of structural differences of the ganglion cell and inner nuclear layers in children with Down syndrome adds materially to the literature regarding the neurological sequelae of Down syndrome, and we hypothesise mechanisms by which these differences may occur.
Summary
What was known before
-
Down syndrome is associated with characteristic changes to the majority of ocular tissues.
-
Abnormalities of visual function are common in children with Down syndrome.
-
Published reports on retinal structure in Down syndrome are variable.
What this study adds
-
Hand-held OCT can be used to characterise retinal architecture in young children with Down syndrome in an outpatient setting.
-
Foveal hypoplasia may be present in the majority of children with Down syndrome.
-
Macular thickening may be due to persistence of the inner retinal layers, particularly the ganglion cell and inner nuclear layers at the fovea.
References
Patterson D. Molecular genetic analysis of Down syndrome. Hum Genet. 2009;126:195–214.
Kazemi M, Salehi M, Kheirollahi M. Down Syndrome: Current Status, Challenges and Future Perspectives. Int J Mol Cell Med [Internet]. 2016;5:125–33.
Weijerman ME, De Winter JP. Clinical practice: The care of children with Down syndrome. Eur J Pediatrics. 2010;169:1445–52.
Cullen JF. Blindness in Mongolism (Down’s Syndrome). Br J Ophthalmol. 1963;44:331–3.
Traboulsi EI, Levine E, Mets MB, Parelhoff ES, O’Neill JF, Gaasterland DE. Infantile glaucoma in down’s syndrome (Trisomy 21). Am J Ophthalmol. 1988;105:389–9.
Stirn Kranjc B. Ocular Abnormalities and Systemic Disease in Down Syndrome. Strabismus [Internet]. 2012;20:74–7.
Kim JH, Hwang JM, Kim HJ, Yu YS. Characteristic ocular findings in Asian children with Down syndrome. Eye. 2002;16:710–4.
Stirn Kranjc B. Ocular abnormalities and systemic disease in down syndrome. In: Strabismus. 2012. 74–7.
Creavin AL, Brown RD. Ophthalmic assessment of children with down syndrome: Is England doing its bit. Strabismus. 2010;18:142–5.
Morton GV. Why do children with down syndrome have subnormal vision? Am Orthopt J 2011;61:60–70.
Stephen E, Dickson J, Kindley AD, Scott CC, Charleton PM. Surveillance of vision and ocular disorders in children with Down syndrome. Dev Med Child Neurol [Internet]. 2007;49:513–5.
John FM, Bromham NR, Woodhouse M, Candy TR. Spatial vision deficits in infants and children with Down syndrome. Investig Ophthalmol Vis Sci. 2004;45:1566–72.
Ljubic A, Trajkovski V, Stankovic B. Strabismus, refractive errors and nystagmus in children and young adults with Down syndrome. Ophthalmic Genet. 2011;32:204–11.
Shapiro MB, France TD. The ocular features of Down’s syndrome. Am J Ophthalmol. 1985;99:659–63.
Felius J, Beauchamp CL, Stager DR. Visual acuity deficits in children with nystagmus and down syndrome. Am J Ophthalmol. 2014;157:458–63.
Caputo AR, Wagner RS, Reynolds DR, Guo S, Goel AK. Down Syndrome: Clinical Review of Ocular Features. Clin Pediatr (Philos). 1989;28:355–8.
Berk AT, Saatci AO, Erçal MD, Tunç M, Ergin M. Ocular findings in 55 patients with Down’s syndrome. Ophthalmic Genet [Internet]. 1996;17:15–9.
Fimiani F, Iovine A, Carelli R, Pansini M, Sebastio G, Magli A. Incidence of ocular pathologies in Italian children with Down syndrome. Eur J Ophthalmol. 2007;17:817–22.
Little JA, McCullough S, McClelland J, Jackson AJ, Saunders KJ. Low-contrast acuity measurement: Does it add value in the visual assessment of down syndrome and cerebral palsy populations? Investig Ophthalmol Vis Sci. 2013;54:251–7.
Little JA, Woodhouse JM, Lauritzen JS, Saunders KJ. Vernier acuity in down syndrome. Investig Ophthalmol Vis Sci. 2009;50:567–72.
Woodhouse JM, Meades JS, Leat SJ, Saunders KJ. Reduced accommodation in children with Down syndrome. Investig Ophthalmol Vis Sci. 1993;34:2382–7
Cregg M, Woodhouse JM, Pakeman VH, Saunders KJ, Gunter HL, Parker M, et al. Accommodation and refractive error in children with Down syndrome: Cross-sectional and longitudinal studies. Investig Ophthalmol Vis Sci. 2001;42:55–63.
Hendrickson A, Possin D, Vajzovic L, Toth CA. Histologic Development of the Human Fovea From Midgestation to Maturity. Am J Ophthalmol. 2012;154:767–78.
Wang J, Spencer R, Leffler JN, Birch EE. Critical period for foveal fine structure in children with regressed retinopathy of prematurity. Retina 2012;32:330–9.
Wallman J, Winawer J. Homeostasis of eye growth and the question of myopia. Neuron. 2004;43:447–68.
Kondo H. Foveal hypoplasia and optical coherence tomographic imaging. Taiwan J Ophthal. 2018;8:181–88.
Patel A, Purohit R, Lee H, Sheth V, Maconachie G, Papageorgiou E, et al. Optic Nerve Head Development in Healthy Infants and Children Using Handheld Spectral-Domain Optical Coherence Tomography. Ophthalmology. 2016;123:2147–57.
Avery RA, Cnaan A, Schuman JS, Chen CL, Glaug NC, Packer RJ, et al. Reproducibility of circumpapillary retinal nerve fiber layer measurements using handheld optical coherence tomography in sedated children. Am J Ophthalmol. 2014;154:780–87.e1.
Tong AY, El-Dairi M, Maldonado RS, Rothman AL, Yuan EL, Stinnett SS, et al. Evaluation of optic nerve development in preterm and term infants using handheld spectral-domain optical coherence tomography. Ophthalmology. 2014;121:1818–26.
Rothman AL, Sevilla MB, Freedman SF, Tong AY, Tai V, Tran-Viet D, et al. Assessment of retinal nerve fiber layer thickness in healthy, full-term neonates. Am J Ophthalmol. 2015;159:803–11.
Hill CM, Evans HJ, Elphick H, Farquhar M, Pickering RM, Kingshott R, et al. Prevalence and predictors of obstructive sleep apnoea in young children with Down syndrome. Sleep Med. 2016;27–28:99–106.
Burfield HJ, Patel NB, Ostrin LA. Ocular biometric diurnal rhythms in emmetropic and myopic adults. Investig Ophthalmol Vis Sci. 2018;59:5176–87.
Chakraborty R, Read SA, Collins MJ. Diurnal variations in axial length, choroidal thickness, intraocular pressure, and ocular biometrics. Investig Ophthalmol Vis Sci. 2011;52:5121–9.
Lee H, Proudlock F, Gottlob I. Is handheld optical coherence tomography reliable in infants and young children with and without nystagmus? Invest Ophthalmol Vis Sci. 2013;54:8152–9.
Thomas MG, Kumar A, Mohammad S, Proudlock FA, Engle EC, Andrews C, et al. Structural grading of foveal hypoplasia using spectral-domain optical coherence tomography: A predictor of visual acuity? Ophthalmology. 2011;118:1653–60.
Laguna A, Barallobre MJ, Marchena MÁ, Mateus C, Ramírez E, Martínez-Cue CC, et al. Triplication of dyrk1a causes retinal structural and functional alterations in down syndrome. Hum Mol Genet. 2013;22:2275–84.
O’Brien S, Wang J, Smith HA, Donaldson DL, Haider KM, Roberts GJ, et al. Macular structural characteristics in children with Down syndrome. Graefe’s Arch Clin Exp Ophthalmol. 2015;253:2317–23.
Mangalesh S, Vinekar A, Jayadev C, Kemmanu V, Bhat M, Sivakumar M, et al. Spectral Domain Optical Coherence Tomography in Detecting Sub-Clinical Retinal Findings in Asian Indian Children with Down Syndrome. Curr Eye Res. 2019;44:901–07.
Nelson LB. Dynamics of Human Foveal Development after Premature Birth. Yearb Ophthalmol. 2012;2012:184–5.
Vajzovic L, Hendrickson AE, O’Connell RV, Clark LA, Tran-Viet D, Possin D, et al. Maturation of the Human Fovea: Correlation of Spectral-Domain Optical Coherence Tomography Findings With Histology. Am J Ophthalmol. 2012;154:779–789. e2
Georges P, Madigan MC, Provis JM. Apoptosis during development of the human retina: Relationship to foveal development and retinal synaptogenesis. J Comp Neurol. 1999;413:198–208.
Thomas MG, Papageorgiou E, Kuht HJ, Gottlob I Normal and abnormal foveal development. Br J Ophthalmol. 2020:316348
Golden JA, Hyman BT. Development of the superior temporal neocortex is anomalous in trisomy 21. J Neuropathol Exp Neurol. 1994;53:513–20.
Engidawork E, Lubec G. Molecular changes in fetal Down syndrome brain. J Neurochem. 2003;84:895–904.
Behar TN, Colton CA. Redox regulation of neuronal migration in a down syndrome model. Free Radic Biol Med [Internet]. 2003;35:566–75.
Huo HQ, Qu ZY, Yuan F, Ma L, Yao L, Xu M, et al. Modeling Down Syndrome with Patient iPSCs Reveals Cellular and Migration Deficits of GABAergic Neurons. Stem Cell Rep. 2018;10:1251–66.
Noval S, Freedman SF, Asrani S, MA. E-D Incidence of fovea plana in normal children. 2014;18:471–5.
Villegas VM, Schwartz SG, Hamet TD, McKeown CA, Capo H, HW F Variable clinical profile of fovea plana in normal children. 2018;49:251–7.
Rufai SR, Thomas MG, Purohit R, Bunce C, Lee H, Proudlock FA, et al. Can Structural Grading of Foveal Hypoplasia Predict Future Vision in Infantile Nystagmus?: A Longitudinal Study. Ophthalmology 2020;127:492–500.
Baburamani AA, Patkee PA, Arichi T, Rutherford MA. New approaches to studying early brain development in Down syndrome. Dev Med Child Neurol. 2019;61:867–79.
Acknowledgements
We thank Jan Sniatecki for his contribution to data collection, and Gift of Sight for providing funding for the handheld OCT.
Author information
Authors and Affiliations
Contributions
R.N. was responsible for performing data collection, extracting and analysing data, and writing the report. D.O. was responsible for performing data collection, extracting, and analysing data. L.F. and L.B. were responsible for coordinating data collection and liaison with participants’ families. C.H. was responsible for study design, identification of the subject cohort, and review of the report. HL was responsible for study design, data analysis and review of the report. All authors approved the final draft of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
CMH has received an educational grant from Flynn Pharma and has acted as paid expert advisor for an international practice consensus panel on use of Slenyto (XR melatonin) from Neurim pharmaceuticals.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Nicholson, R., Osborne, D., Fairhead, L. et al. Segmentation of the foveal and parafoveal retinal architecture using handheld spectral-domain optical coherence tomography in children with Down syndrome. Eye 36, 963–968 (2022). https://doi.org/10.1038/s41433-021-01883-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41433-021-01883-6
