
Abstract
Purpose
Tissue inhibitor of matrix metalloproteinase (TIMP)-3 has many functions, including preventing the constituent formation of tumor necrosis factor-alpha (TNFα) in tissue. Sorsby macular dystrophy is caused by a mutation in the gene responsible for TIMP-3, suggesting a potential treatment.
Methods
Comprehensive ophthalmologic examination with multimodal imaging to include optical coherence tomography (OCT) and OCT angiography were used to evaluate a patient with Sorsby fundus dystrophy treated first with intravitreal triamcinolone, then with adalimumab.
Results
A 35-year-old woman presented in 2003 with aggressive macular neovascularization in both eyes related to Sorsby macular dystrophy c.610A>T (p.Ser204Cys). Her visual acuity was 20/25 in the right and 20/400 in the left eye. She was treated with periodic intravitreal injections of 4 mg triamcinolone, which caused the neovascularization to become inactive. When switched to intravitreal bevacizumab, she showed disease activity. She was switched back to intravitreal triamcinolone with minimal signs of exudation and hemorrhage. Because of the high lifetime risk of complication, she was switched to subcutaneous adalimumab and in follow-up over 18 months had no signs of disease activity. The visual acuity in the right eye was 20/20.
Conclusions
TIMP3 has numerous effects including controlling local TNFα production. It is possible with the mutation in the gene for TIMP-3, abnormally high tissue levels of TNFα are produced in the eye. Direct inhibition of TNFα action by adalimumab offers a molecularly targeted approach to the disease pathophysiology and merits increased study.
Similar content being viewed by others

Sorsby fundus dystrophy was first described in 1949 in five families that showed a dominant inheritance pattern of macular lesions, which in early stages demonstrated “oedema and hemorrhagic-exudative reaction in the central areas” [1, 2]. Eventually, the eyes developed an appearance that “suggest a central choroiditis that has run its course” and the fundus developed choroidal sclerosis [1]. The patients were affected by about 40 years of age [1]. Although Sorsby, and others, commented the disease appeared to have an inflammatory basis, Duke-Elder referred to the condition as Sorsby’s pseudo-inflammatory fundus dystrophy, because he doubted the role of inflammation [3]. In 1994, Weber et al. [4] found that patients with Sorsby fundus dystrophy have a mutation in the tissue inhibitor of metalloproteinases-3 (TIMP-3), one of a family of four enzymes classified by their ability to inhibit matrix metalloproteinases (MMPs) [2]. TIMP-3 has been found to have many functions in addition to inhibiting classic MMPs, one of which is to inhibit a disintegrin and metalloproteinase 17 (ADAM17) [5], which is responsible for the constitutive secretion of tumor necrosis factor-alpha (TNFα). TIMP-3 is the only TIMP that blocks ADAM17, also known as TNFα converting enzyme [6]. Excessive secretion of TNFα has been associated with tissue damage and angiogenesis [7]. A patient with Sorsby macular dystrophy being treated with quarterly intravitreal injections of triamcinolone retained central vision for many years, but eventually required cataract extraction and filtering surgery [8]. Because of the high lifetime risk for endophthalmitis from the intravitreal injections, she was switched to adalimumab and her disease activity ceased for the follow-up period, now 18 months later. She has not required any rescue therapy with corticosteroids or anti-VEGF therapy.
Case report
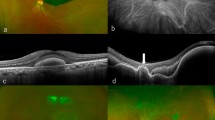
A 35-year-old woman presented in 2003 with a history of having PDT in the right eye once, and five times in the left for bilateral macular neovascularization. Her visual acuity was 20/25 in the right and 20/400 in the left. She had a family history of macular neovascularization starting at an early age in her mother, maternal aunt, and two maternal cousins. Her family hailed from England and Scotland. The patient had a disease causing TIMP-3 mutation, c.610A>T (p.Ser204Cys). The patient was treated with PDT and an intravitreal injection of triamcinolone 4 mg. There was a dramatic reduction in the neovascularization in both eyes. Her right eye showed signs of recurrence of the exudation from the neovascularization 9 months after the PDT with triamcinolone and was retreated in a similar manner. Her left eye had extensive scarring and received no further treatment. Thereafter, she was treated with periodic intravitreal triamcinolone injections alone [8]. The patient was switched to bevacizumab and later with ranibizumab, but with each of these she repeatedly developed signs of exudation and hemorrhage by 1 month after injection. Therefore, the patient was maintained on intravitreal injections of 4 mg triamcinolone approximately every 3–4 months. She required a cataract extraction for a corticosteroid-related cataract and filtering surgery, for uncontrolled glaucoma. Optical coherence tomography showed an extensive double-layered sign with optical coherence tomography revealing the patient developed a large area of neovascularization (Fig. 1).

A After 17 years of corticosteroid treatment she had a widespread double-layer sign between the retinal pigment epithelium (green arrows) and Bruch’s membrane (yellow arrows) but retained good visual acuity. B Widespread macular neovascularization was confirmed in this 9 × 9 mm area imaged using optical coherence tomography angiography. C During the corticosteroid treatment period the patient occasionally developed small areas of hemorrhage (white arrows) and lipid exudation (yellow arrows) at the outer edge of the neovascularization. D The patient was switched to twice monthly adalimumab treatment. After 18 months she had no evidence of exudation or hemorrhage. E The autofluorescence imaging shows an intact retinal pigment epithelium except for a small area of atrophy (arrow), which was seen in the earlier optical coherence tomography scan as hypertransmission (A).
Because of the perceived increased risk for endophthalmitis in an eye with a filtering bleb receiving intraocular corticosteroid injections, an effort was made to switch the patient to a therapy with a lower endophthalmitis risk. Given the role TIMP-3 has in the inhibition of TNFα production, a TNFα inhibitor, adalimumab, was started with two 40 mg doses subcutaneously followed by 40 mg every twice monthly. The patient showed a complete cessation of disease activity and did not need either corticosteroid or anti-VEGF rescue therapy. She has had no known side-effects while using adalimumab.
Discussion
The underlying problem in Sorsby macular dystrophy is a mutation in TIMP-3 [4]. The accumulation of extracellular material, with progression to neovascularization and atrophy from a mutation in an inhibitor of MMPs, enzymes that function to breakdown the extracellular matrix, led to the formation of many theories over the years. Loss of function from the mutation would lead to unrestricted activity of MMPs, which intuitively would not seem to explain the increase in extracellular material deposition. Gain of inhibitory function of the mutated TIMP through persistent dimer formation could potentially explain some of the findings, except normal TIMP-3 forms dimers to the same degree as the TIMP-3 mutation seen in Sorsby macular dystrophy [9]. TIMP-3 affects VEGF binding to VEGF receptor 2 [10], although how this leads to neovascularization, scarring, atrophy, and choroidal thinning is not evident.
TIMP-3 inhibits ADAM17, which cleaves the transmembrane precursors of multiple cytokines, growth factors, and receptors. Following cleavage there is a release of a soluble ectodomain, in a process known as shedding. ADAM17 constitutively converts a transmembrane precursor to TNFα, which is free to diffuse into tissue. TNFα is a powerful pro-inflammatory mediator and can induce angiogenesis [7]. ADAM17 is inhibited by TIMP-3. Deletion of mouse TIMP-3 results in excessive TNFα levels and early death because of lung and liver problems [11]. It is possible that TIMP-3 mutation in Sorsby macular dystrophy leads to a loss in ADAM17 inhibition. Other mutations of TIMP-3 are associated with eye disease and systemic findings in humans. For example, c.572A>G (p.Y191C) is associated with severe emphysema and c.113C>G, p.S38C is associated with bronchiectasis in addition to the eye disease [12]. It is possible that various phenotypes of disease affecting the eye and elsewhere are associated with specific mutations of TIMP-3. The recognition of the c.610A>T (p.Ser204Cys) mutation by ophthalmologists may rest in the preferential ocular involvement caused by this mutation.
TNFα induces cultured retinal pigment epithelial cells to secrete proteins relevant to the pathogenesis of age-related macular degeneration (AMD), including complement factor 3 [13]. Impeding the complement system, including complement factor 3, has been the focus of several investigations into inhibiting progression of AMD [14]. Increased complement cascade activation, related to specific complement factor H (CFH) polymorphisms, has been associated with drusen nucleation, accumulation of material in drusen, and manifestations of late AMD [15]. Eyes with Sorsby macular dystrophy also have accumulation of deposits in the fundus similar to those found in AMD, but instead of specific CFH polymorphisms, it is possible that the accumulations may be initiated by increased local levels of TNFα. Complement directed attack on the choriocapillaris increases with age and is prominent in AMD [14]. Choriocapillaris abnormalities have been reported in eyes with Sorsby macular dystrophy [8].
Inhibiting TNFα using biologics such as adalimumab has transformed the treatment of many diseases including plaque psoriasis, psoriatic arthritis, rheumatoid arthritis, juvenile rheumatoid arthritis, Crohn’s disease, and ankylosing spondylitis to name a few. These diseases have pathophysiologic pathways that eventually involve the production of excessive amounts of TNFα. Inhibiting the effects of TNFα can lead to amelioration of disease without necessarily addressing the upstream cause of the TNFα secretion. In the Sorsby macular dystrophy, adalimumab may not only inhibit these downstream inflammatory cascades but may also directly inhibit what could be a major initiator of disease, primary overproduction of TNFα. As such, anti-TNFα agents may be a molecularly targeted treatment of Sorsby fundus dystrophy. Conversion to more aggressive forms of Sorsby fundus dystrophy may occur from the combined effects of elevated TNFα and changes such as Bruch’s membrane deposits. It may be desirable to treat patients early in the course of disease to avoid all potential TNFα-related changes, thereby avoiding the later phases of disease. This could involve treating patients discovered to have TIMP-3 mutations based on screening driven by known family histories with institution of treatment at an age prior to those known to be associated with the devastating consequences of Sorsby fundus dystrophy. This could obviate need to simultaneously manage the potentially damaging effects of both Sorsby fundus dystrophy and those related to neovascularization.
This case report illustrates only one treated patient, thus having the weakness of a small sample size. There appears to be varying levels of disease severity reported in the literature, with some patients being successfully managed with sporadic injections of anti-VEGF agents on one end of the spectrum to the present patient, who seemed to have a poor response to anti-vascular endothelial growth factor drugs. Treatment with adalimumab may address an underlying pathogenic mechanism of Sorsby fundus dystrophy and may obviate the need for anti-vascular endotheial growth factor injections.
Summary
What was known before
-
Sorsby fundus dystrophy is caused by a mutation in the gene encoding tissue inhibitor of metalloproteinases-3 (TIMP-3).
-
TIMP-3 has many functions. One of the functions of TIMP-3 is to inhibit a disintegrin and metalloproteinase 17 (ADAM17).
-
The only TIMP that inhibits ADAM17 is TIMP-3.
-
ADAM17 causes the production and release of tissue necrosis factor alpha (TNFα).
What this study adds
-
A patient was treated with adalimumab, a biologic that binds to and inactivates TNFα.
-
Following initiation of treatment with adalimumab, disease activity showed cessation for the 18 months of current follow-up.
-
Adalimumab may offer a molecularly targeted treatment of Sorsby fundus dystrophy.
References
Sorsby A, Mason MEJ. A fundus dystrophy with unusual features. Br J Ophthalmol. 1949;33:67–97.
Capon MRC, et al. Sorsby’s fundus dystrophy: a light and electron microscopic study. Ophthalmology. 1989;96:1769–77.
Duke-Elder S, Perkins ES. Diseases of the uveal tract. System of ophthalmology. London: Kimpton; 1966.
Weber BHF, Vogt G, Pruett RC, Stöhr H, Felbor U. Mutations in the tissue inhibitor of metalloproteinases-3 (TIMP3) in patients with Sorsby’s fundus dystrophy. Nat Genet. 1994;8:352–6.
Kawai T, Elliott KJ, Scalia R, Eguchi S. Contribution of ADAM17 and related ADAMs in cardiovascular diseases. Cell Mol Life Sci. 2021;78:4161–87.
Smookler DS, et al. Cutting edge: tissue inhibitor of metalloproteinase 3 regulates TNF-dependent systemic inflammation. J Immunol. 2006;176:721–5.
Kalliolias GD, Ivashkiv LB. TNF biology, pathogenic mechanisms and emerging therapeutic strategies. Nat Rev Rheumatol. 2016;12:49–62.
Spaide RF. Long-term visual acuity preservation in Sorsby fundus dystrophy with corticosteroid treatment. Retin Cases Br Rep. 2019. https://doi.org/10.1097/ICB.0000000000000946.
Hongisto H, et al. In vitro stem cell modelling demonstrates a proof-of-concept for excess functional mutant TIMP3 as the cause of Sorsby fundus dystrophy. J Pathol. 2020;252:138–50.
Qi JH, et al. A novel function for tissue inhibitor of metalloproteinases-3 (TIMP3): inhibition of angiogenesis by blockage of VEGF binding to VEGF receptor-2. Nat Med. 2003;9:407–15.
Mohammed FF, et al. Abnormal TNF activity in Timp3−/− mice leads to chronic hepatic inflammation and failure of liver regeneration. Nat Genet. 2004;36:969–77.
Meunier I, et al. A new autosomal dominant eye and lung syndrome linked to mutations in TIMP3 gene. Sci Rep. 2016;6:1–9.
An E, Gordish-Dressman H, Hathout Y. Effect of TNF-alpha on human ARPE-19-secreted proteins. Mol Vis. 2008;14:2292–303.
Wu J, Sun X. Complement system and age-related macular degeneration: drugs and challenges. Drug Des Dev Ther. 2019;13:2413–25.
Hageman GS, et al. An integrated hypothesis that considers drusen as biomarkers of immune-mediated processes at the RPE-Bruch’s membrane interface in aging and age-related macular degeneration. Prog Retinal Eye Res. 2001;20:705–32.
Acknowledgements
The study was funded in part by the Macula Foundation, Inc., which had no input in terms of content.
Funding
Topcon Medical Systems, Regeneron, Roche, Genentech, Heidelberg Engineering, Adverum Biotechnologies, and DORC.
Author information
Authors and Affiliations
Contributions
This is a single author study.
Corresponding author
Ethics declarations
Competing interests
The author declares no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Spaide, R.F. Treatment of Sorsby fundus dystrophy with anti-tumor necrosis factor-alpha medication. Eye 36, 1810–1812 (2022). https://doi.org/10.1038/s41433-021-01735-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41433-021-01735-3
This article is cited by
-
Bevacizumab/ranibizumab/triamcinolone
Reactions Weekly (2021)
