
Abstract
Background/aims
A Bitot spot is a conjunctival lesion, classically associated with severe vitamin A deficiency. In this paediatric series, we describe conjunctival lesions indistinguishable from Bitot spots, seen in the presence of normal vitamin A levels.
Methods
This descriptive case series was performed by retrospective review of case notes, including all patients with Bitot-like spots found to have normal serum vitamin A levels, seen at the Hospital for Sick Children, Toronto, between 2006 and 2016. Data collected included age at presentation, ophthalmic and systemic diagnoses, and the presence of recognised genetic mutations. Histopathology was reviewed in one case.
Results
Ten patients with Bitot-like spots with laboratory-confirmed normal serum vitamin A levels were identified. The conjunctival lesions were indistinguishable clinically and histopathologically from classic Bitot spots and were noted to occur in a range of anterior segment pathologies, including aniridia, WAGR syndrome, Axenfeld–Rieger syndrome, and blepharokeratoconjunctivitis.
Conclusions
Bitot-like spots are found in children with a number of anterior segment pathologies in the absence of vitamin A deficiency.
Similar content being viewed by others

Introduction
A Bitot spot, first described by Pierre Bitot in 1863 [1, 2], is a conjunctival lesion classically associated with xerophthalmia, an umbrella term referring to the ocular manifestations of vitamin A deficiency [3]. Clinically, these lesions appear as well-defined, foamy white plaques on the bulbar conjunctiva. They are most commonly round or triangular in shape [2, 4], with a base abutting the temporal limbus and an apex projecting towards the lateral canthus. Extensive involvement of the bulbar conjunctiva is less common [2]. Appearance and incidence does not vary between adults and children [2].
The histopathology of a Bitot spot reveals metaplastic squamous epithelium together with keratin tangles and a loss of goblet cells. Gas-producing bacteria, Corynebacterium xerosis [5], dwell in the conjunctiva creating pockets of gas, giving the lesion its distinct foamy appearance [5, 6].
In the past, Bitot spots were considered to be pathognomic of vitamin A deficiency [5], allowing their presence to be used for screening patients for xerophthalmia, a preventable cause of blindness in the developing world. However, these lesions are also seen in other vitamin deficiencies, such as pellagra, a vitamin B deficiency state, as well as in the absence of any identifiable nutritional deficiency [2, 7]. The prevalence of cases reported from hot climates have led to the suggestion that exposure, drying, and actinic stimulation may play a role in their creation [7,8,9,10].
Here we present a paediatric case series of lesions clinically indistinguishable from the classic Bitot spot in the presence of normal vitamin A levels, almost all of which were associated with congenital or acquired anterior segment pathologies.
Methods
This study was approved by the Hospital for Sick Children Research Ethics Board.
Ten consecutive cases were included with consent, all of whom were diagnosed with Bitot-like spots from April 2006 to November 2016, and in all of whom normal serum vitamin A levels were found on testing. Retrospective chart review was used to document date and age at diagnosis, visual acuities, other diagnosed ophthalmic and systemic conditions, and topical medication use, as well as any positive results from genetic panels.
All patients had clinical slit lamp photographs of their lesion, which were reviewed. The shape and size of the lesion, laterality, proximity to the limbus, orientation, and whether it had a foamy appearance were noted. In addition, a single patient underwent tissue biopsy of their lesion at the time of glaucoma seton surgery, which was sent for histopathological examination.
Results
Ten children with a mean age at presentation of 7.9 years (range 3–11 years) were included and their findings are summarised in Table 1. Serum vitamin A levels ranged from 0.9 to 2.7 μmol/L, with 0.52 μmol/L being the lower limit of the normal range.
Four patients had single, unilateral lesions only, while six were bilaterally affected. Patients 2 and 7, both of whom had WAGR (Wilms tumour, Aniridia, Genitourinary abnormalities, Range of developmental delays) with very similar genetic anomalies had lesions present both nasally and temporally in the same eye, representing more extensive disease in terms of surface area.
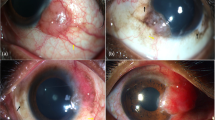
Lesions were predominantly seen in the inferotemporal quadrant, and only one patient had a unilateral nasal lesion. All lesions were seen at a maximum of 4 mm away from the limbus and found to be oval or triangular in all cases except one more extensive polyhedral lesion. Triangular lesions had bases facing the limbus in all cases. In all, 50% of lesions were foamy. Images are shown in Fig. 1.

A Inferotemporal lesion with foamy appearance in right eye of patient 1. B Nasal lesion in right eye of patient 3. C Close-up image of foamy appearance of lesion in patient 4. D Nasal lesion in right eye of patient 9.
All cases except one had an ophthalmic diagnosis affecting the anterior segment of the eye. Four patients had aniridia. In one patient this was sporadic, and in three of the four this was part of WAGR syndrome. Similarly, a patient with foveal hypoplasia was found to have a variant of unknown significance in the PAX6 gene. Two patients had Axenfeld–Rieger syndrome with confirmed mutations in the FOXC1 gene. One patient had microphthalmos and X-linked Nance–Horan syndrome. Another patient had inferior pannus of the cornea caused by blepharokeratoconjunctivitis with no systemic diagnosis, and a final patient had otherwise normal anterior segment appearance. In these 2 patients, genetic testing was not carried out.
Six patients had glaucoma, all of whom were on topical treatment with combination carbonic anhydrase inhibitor/timolol eyedrops, and three of the six were using apraclonidine and a prostaglandin analogue in addition. Five patients had prolonged (≥2 years) exposure to preserved topical medications at the time of diagnosis.
Patients in whom unilateral (4) versus bilateral (6) lesions were present were compared. Results are displayed in Table 2. PAX6 mutations were seen in 25% of unilateral cases compared to 67% of bilateral cases. The mean number of eye drops used was also found to be higher in the bilateral group at 2.3, compared to 1.3 in the unilateral group. There was no difference between the groups in previous exposure to preserved medications.
An incisional biopsy was taken from one patient in the region of the Bitot-like spot, which was sent for histopathological examination. This revealed fibrovascular tissue covered with keratinising squamous epithelium and a localised focus of subepithelial chronic inflammation (see Fig. 2).

The scale bar represents 300 microns.
Discussion
This case series reports conjunctival lesions, indistinguishable clinically from Bitot spots, in children with normal serum vitamin A levels, in the presence of a number of anterior segment pathologies or normal ocular examination.
Compared to the description of the classic Bitot spot in vitamin A deficiency [1, 2, 5], no difference was noted in the presentation of lesions in terms of shape, proximity to the limbus, or location. However, some features typical of classic Bitot spot were not seen. Only 60% of lesions were bilateral in this series, compared to 95% in vitamin A deficiency [2], and the characteristic foamy appearance was seen in only half the lesions.
The classic histopathological changes of Bitot spots include irregular maturational sequence and disorganised basal layers of conjunctival epithelium with keratinisation of the surface layers, in association with inflammatory infiltration of conjunctival substantia propria and loss of goblet cells [5]. Histopathological examination of one lesion in this series revealed the same features, indicating no difference from Bitot spots seen in vitamin A deficiency.
Although the exact mechanism by which Bitot spots form is unknown, their presence in vitamin A deficiency may indicate that this vitamin is required for normal metabolism of conjunctival epithelium. Of note, observed tissue changes in Bitot spots respond within 7 days of commencing high-dose vitamin A (200,000 IU orally) and goblet cells return within 2 weeks [11]. Not all lesions respond to high-dose vitamin A supplementation therapy however, and although resistant lesions are not clinically distinct, histopathologically they are more defined, resulting from persistent metaplastic change [2, 11, 12]. Considering this, previous undiagnosed vitamin A deficiency is a possible cause of lesions and is recognised as a limitation of this retrospective study, which only accounted for present vitamin A levels, and relies on absence of a reported history of vitamin deficiency.
Notably, genetic testing revealed that half (5/10) the number of patients in this series had PAX6 mutations, 4 in association with aniridia, and a further case had a PAX6 mutation of unknown significance. PAX6 is a transcription factor that acts in embryonic development of the eye and brain. Indeed, limbal stem cell deficiency is a feature of aniridia with metaplasia of these cells demonstrable on impression cytology [13]. Likewise, two patients have mutations in the FOXC1 gene, which is also a transcription factor gene involved in anterior segment and glaucoma phenotypes. Both genes could play a role in normal conjunctival metabolism.
Other suggested mechanisms of how these lesions form include local trauma from thermal or chemical injury [7, 14]. Most patients in this series were on intraocular pressure-lowering medications, prolonged use of which, particularly in the context of benzalkonium chloride (BAK) preservatives, can cause tear film instability, conjunctival inflammatory changes, and corneal epitheliopathy, as well as squamous metaplasia and loss of conjunctival goblet cells [15]. Our patients were managed with preservative-free medications where possible, but previous exposure to preservatives did occur as detailed in Table 1. Half the number of the patients in the series had prolonged exposure to one or more preserved medications. Patient 3 did not undergo genetic testing but had evidence of previous ocular surface inflammation and use of BAK-preserved lubricants, which could be relevant. Patient 10 had no identified anterior segment pathology or topical medication use, and hence the mechanism of Bitot-like spot formation is unknown. No other medical problems were identified, but an unrecognised history of nutritional deficiency cannot be excluded.
Comparing cases in which unilateral or bilateral lesions occurred, it is evident that patients with PAX6 mutations and WT1 deletions predominantly had bilateral lesions. Likely due to associated genetic phenotypes, the mean best corrected visual acuity was lower in this group, secondary to corneal pathology and glaucoma. These pathologies could also explain why the mean eyedrop use in the bilateral group was higher than the unilateral group. No specific association is seen between bilaterality and prolonged preserved medication use, as 50% of cases in each group had exposure to preserved medications.
In the absence of vitamin A deficiency, this series indicates association of anomalies in anterior segment genes, principally PAX6, and perhaps also use of topical medications with occurrence of Bitot-like spots. The interdependency of these genetic and environmental factors makes it purely speculative which factor is of greater importance.
Interestingly in the developed world, Bitot spot in children is increasingly seen in the context of vitamin A deficiency secondary to restricted diets adopted by patients with behavioural disorders, including autism [16, 17]. Their presence should therefore still alert the clinician to the possibility of malnutrition and associated xerophthalmia. However, in cases where serum vitamin A levels are normal, there may be an association with other anterior segment pathologies, and stigmata of these should be looked for. Further, given that mutations in anterior segment genes were identified in a high proportion of patients in this study, consideration should be given to testing for appropriate anterior segment gene panels in cases where presence of lesions is unexplained.
Summary
What was known before
-
Bitot’s spot is a conjunctival lesion, once thought to be pathognomic of vitamin A deficiency, and used for recognising the possible retinal dysfunction seen in xerophthalmia.
-
These lesions have been recognised to occur in normal vitamin A levels, but no recent or robust data exist on the nature of their occurrence.
What this study adds
-
Lesions indistinguishable clinically and histopathologically from the classic Bitot spot are seen children with normal vitamin A levels.
-
Their occurrence has been noted predominantly in children with anterior segment pathologies, including genetic defects notably in association with PAX6 mutations, which may shed light on the relevant factors in their creation.
References
Bitot C. Gaz hebd. Med Chir. 1863;10:284.
Rodger FC, Saiduzzafar H, Grover AD, Fazal A. A reappraisal of the ocular lesion known as the Bitot’s spot. Br J Nutr. 1963;17:475.
Sommer A, World Health Organization. Vitamin A deficiency and its consequences: a field guide to detection and control. 3rd ed. Geneva: World Health Organization; 1995.
Shukla M, Behari K. Congenital Bitot spots. Indian J Ophthalmol. 1979;27:63–4.
Sommer A. Xerophthalmia and vitamin A status. Prog Retinal Eye Res. 1998;17:9–31.
Ferrari G, Vigano M. Bitot’s spot in vitamin A deficiency. NEJM 2013;368:e29.
Levine RA, Rabb MF. Bitot’s spot overlying a pinguecula. Arch Ophthal. 1971;86:525–8.
Paton D, McClaren DS. Bitot spots. Am J Ophthalmol. 1960;50:568.
Aykroyd WR, Rajagopal K. The state of nutrition of children in south India. Indian J Med Res. 1936;24:419–37.
Nickolls L, Nimalasuriya A. Bitot’s spots in Ceylon. Lancet. 1939;236:1432.
Sommer A. Bitot’s spots responsive and unresponsive to vitamin A. Arch Ophthalmol. 1981;99:2014–27.
Djunaedi E, Sommer A, Pandji A, Kusdiono, Taylor HR. Impact of vitamin A supplementation on xerophthalmia: a randomised controlled community trial. Arch Ophthalmol. 1988;106:218–22.
Jastaneiah S, Al-Rajhi A. Association of aniridia and dry eyes. Ophthalmology. 2005;112:1535–40.
Krishna U, Kamath SJ, Nayak M. Management of Bitot’s spots, cornea ophthalmic pearls. Eyenet Mag. 2016;35–6.
Baudouin C, Labbe A, Liang H, Pauly A, Brignole-Baudouin F. Preservatives in eyedrops: the good, the bad and the ugly. Prog Retin Eye Res. 2010;29:312–34.
Chiu M, Dillon A, Watson S. Vitamin A deficiency and xerophthalmia in children of the developed world. J Paediatr Child Health. 2016;52:699–703.
Chiu M, Watson S. Xerophthalmia and vitamin A deficiency in an autistic child with a restricted diet. BMJ Case Rep. 2015;2015:bcr2015209413.
Acknowledgements
Many thanks to Cynthia VandenHoven and Leslie MacKeen for photo collation and editing.
Author information
Authors and Affiliations
Contributions
AM collected data and wrote and revised the paper. DR wrote an early draft of the paper. MSK and AS collected data. L-NH reported histopathological findings. MDR edited the paper. NN-T contributed patients to the study. AA conceived the idea of the study, contributed patients, collected data, and oversaw writing and editing of the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Maudgil, A., Rachdan, D., Khan, M.S. et al. Bitot-like spots in children with normal vitamin A levels. Eye 36, 1896–1899 (2022). https://doi.org/10.1038/s41433-021-01569-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41433-021-01569-z
