
Abstract
Background
Cataract surgery is the most common operation performed worldwide. A fixed topical corticosteroid-antibiotic combination is usually prescribed in clinical practice for 2 or more weeks to treat post surgical inflammation and prevent infection. However, this protracted schedule may increase the incidence of corticosteroid-related adverse events and notably promote antibiotic resistance.
Methods
This International, multicentre, randomized, blinded-assessor, parallel-group clinical study evaluated the non-inferiority of 1-week levofloxacin/dexamethasone eye drops, followed by 1-week dexamethasone alone, vs. 2-week gold-standard tobramycin/dexamethasone (one drop QID for all schedules) to prevent and treat ocular inflammation and prevent infection after uncomplicated cataract surgery. Non-inferiority was defined as the lower limit of the 95% confidence interval (CI) around a treatment difference >–10%. The study randomized 808 patients enrolled in 53 centres (Italy, Germany, Spain and Russia). The primary endpoint was the proportion of patients without anterior chamber inflammation on day 15 defined as the end of treatment. Endophthalmitis was the key secondary endpoint. This study is registered with EudraCT code: 2018-000286-36.
Results
After the end of treatment, 95.2% of the patients in the test arm vs. 94.9% of the control arm had no signs of inflammation in the anterior chamber (difference between proportions of patients = 0.028; 95% CI: −0.0275/0.0331). No case of endophthalmitis was reported. No statistically significant difference was evident in any of the other secondary endpoints. Both treatments were well tolerated.
Conclusions
Non-inferiority of the new short pharmacological strategy was proven. One week of levofloxacin/dexamethasone prevents infection, ensures complete control of inflammation in almost all patients and may contain antibiotic resistance.
Similar content being viewed by others


Introduction
Cataract is the leading cause of blindness [1] and cataract surgery may be considered the most common surgical intervention worldwide [2,3,4]. As corneal incision is very small (3 mm or less), stitches are unnecessary and surgical wound healing generally occurs spontaneously in <7 days. Following cataract surgery, patients are usually treated to control the post surgical inflammation and prevent infectious complications, frequently with a fixed topical corticosteroid-antibiotic combination [5].
Topical corticosteroids control the ocular inflammation and may prevent cystoid macular oedema [6]. However, the treatment duration is not standardized. Ophthalmologists usually prescribe 2 or more weeks of therapy, often tapering the dose. In this regard, shortening corticosteroid treatment is convenient as the adverse reactions, including intraocular hypertension, are dose dependent [7,8,9].
Regarding the infection prophylaxis, the European Society of Cataract and Refractive Surgeons (ESCRS) recommends the intracameral administration of cefuroxime. However, ocular infection may occur at any time until complete closure of the surgical wound [10,11,12] and for this reason topical administration of antibiotic, although discretionary, is frequently prescribed [13]. The duration of topical prophylaxis is also not standardized and in clinical practice, is generally continued for at least 2 weeks. To prolong the topical antibiotic beyond wound recovery has no convincing justification [11, 12], may be useless, and could promote bacterial resistance [13,14,15,16,17,18].
Based on these considerations, the current study tested the hypothesis that, after uncomplicated cataract surgery, treatment with an antibiotic-corticosteroid combination is required for only 1 week, at least in the large majority of patients.
Materials and methods
This was an international, multicentre, phase III, randomized, blinded-assessor, parallel-group clinical study that evaluated the non-inferiority of the levofloxacin + dexamethasone (L-DSP) treatment (levofloxacin 5 mg/mL + dexamethasone 1 mg/mL) eye drops for 7 days, followed by dexamethasone eye drops (Maxidex®) for an additional 7 days vs. a standard treatment (tobramycin 3 mg/mL + dexamethasone 1 mg/mL, Tobradex®) eye drops for 14 days.
Trial population
The study was conducted in 53 centres located in Italy, Germany, Spain and Russia. Patients were eligible for inclusion if they had had senile or presenile uncomplicated cataract. Exclusion criteria were: (a) ocular conditions that, at the discretion of the investigator, could have interfered with the efficacy and/or safety evaluations (e.g., ocular herpes, blepharitis, conjunctivitis, uveitis, keratitis, diabetic retinopathy, retinal vein occlusions, retinal vasculitis, retinal angiomatous proliferation, pseudo-exfoliation syndrome, intraoperative floppy iris syndrome, etc.); (b) bilateral cataract surgery; (c) treatment with prostaglandin analogues or intravitreal injections of anti-VEGF (vascular endothelial growth factor) drugs; (d) systemic diseases that could have interfered with the results of the study (e.g., rheumatoid arthritis, Sjögren’s syndrome, Behçet’s disease, systemic lupus erythematosus, scleroderma with major ocular involvement, etc.); (e) any condition that could have interfered with the correct instillation of eye drops; (f) ocular surgery in the study eye (including laser surgery) in the 3 months before screening; (g) monocular patients; (h) visual acuity (VA) < 20/80 in the contralateral eye; (i) contraindication to ocular treatment with levofloxacin, tobramycin or dexamethasone; (j) pregnancy or breastfeeding.
Trial information was given in verbal and written formats in the local languages. Written informed consent was provided by all patients before randomization. The protocol was approved by the Ethics Committees of all centres.
Trial groups
Patients were randomized to one of two treatment groups in a 1:1 ratio through an Interactive Web Response System. Randomization was stratified by centre. Treatment began immediately after surgery or after removal of the eye dressing. Doses administered on the same day as surgery (day 0) were considered additional to the full protocol dosage regimen. As shown in Fig. S1, control visits were performed after 3, 7 and 14 days of therapy (day 4, day 8 and day 15, end of the study, respectively) to evaluate the time course of efficacy, compliance, safety and tolerability.
The test arm eye drops (L-DSP) were prescribed for 7 days, followed by dexamethasone eye drops alone (Maxidex®) for an additional 7 days. The control arm was tobramycin + dexamethasone eye drops (Tobradex®) for 14 days. For all eye drops the dosage was one drop—four times a day (hours 8.00, 13.00, 18.00 and 23.00 ± 30 min).
Patients recorded times of instillation in a diary, which was collected to evaluate compliance with treatment. Physician in charge of assessing study parameters was blinded to treatment assignment (blinded assessor) and had no access to the randomization page of the eCRF. A separate unblinded staff was responsible for assigning and dispensing/return study products. Study treatment was dispensed upon randomization (day 0) and on day 8.
Outcome measures
The primary outcome was the proportion of patients without signs of anterior ocular chamber inflammation (sum of cells and flare score = 0) assessed by slit lamp examination after 14 days of treatment (day 15). Cells in the anterior chamber were scored as follows: 0 = no cells; 1 = 1–5 cells; 2 = 6–15 cells; 3 = 16–30 cells; 4 = >30 cells [19]. Aqueous flare (Tyndall effect) was scored: 0 = absent; 1 = trace barely detectable; 2 = mild intensity (iris and lens details clear); 3 = moderate intensity (iris and lens details not clear); 4 = strong intensity (iris and lens details not visible and fibrin in the anterior chamber) [20].
The secondary efficacy outcomes were: (a) incidence of endophthalmitis; (b) proportion of patients without signs of anterior ocular chamber inflammation after 3 and 7 days of treatment; (c) conjunctival hyperaemia (scored: 0 = absence of inflammation, 1 = mild inflammation, i.e. some vessels injected, 2 = moderate inflammation, i.e. diffuse injection of vessels, but individual vessels still discernable, 3 = severe inflammation, i.e. intense injection of vessels, individual vessels not easily discernable) [21] after 3, 7 and 14 days of treatment; (d) Total Ocular Symptoms Score (TOSS); (e) patient-reported evaluation of three ocular symptoms: itching/burning, hyperaemia of conjunctiva and tearing (scored: 0 = none, 1 = mild, 2 = moderate, 3 = severe) [22] after 3, 7 and 14 days of treatment; (f) ocular pain/discomfort (scored: 0 = absent, 1 = mild, 2 = moderate, 3 = severe) after 3, 7 and 14 days of treatment; and (g) use of rescue therapy during treatment.
The safety outcomes were intraocular pressure (IOP), VA analyzed using the decimal unit and adverse events.
The tolerability outcomes were: (a) global evaluation on a four-point scale (0 = no intolerability, 1 = mild intolerability, 2 = moderate intolerability, 3 = maximum intolerability) and (b) ocular complaints of burning, stinging and blurred vision on a four-point scale (0 = none, 1 = mild, 2 = moderate, 3 = severe).
Compliance was assessed through the patient diary.
Statistical analysis
The study planned to enrol 800 patients, 400 in each treatment group. This sample size, adjusted for an expected dropout rate of about 10% [23], would have provided 80% power to assess the non-inferiority of the test treatment compared with the standard therapy at the one-sided 0.025 significance level. This sample size calculation assumed identical 64% success rates (i.e., the proportion of patients without signs of anterior chamber inflammation) in the treatment groups after 14 days of treatment. The success rates assumed are conservative as they correspond to those observed with a less active surface corticosteroid compared with dexamethasone [24]. Non-inferiority was defined as the lower limit of the 95% confidence interval (CI) around a treatment difference >−10%. Based on the literature data, a success rate attributable to a putative placebo is ~30% [25, 26]. Therefore, considering an expected success rate of the test treatment of about 64% and a non-inferiority margin of −10%, the indirect difference between the test treatment and the putative placebo would be of about 25%. This difference was judged as clinically relevant.
Efficacy analyses were performed on the full analysis set (FAS), defined as all randomized patients who applied at least one dose of study treatment. The per protocol set, defined as all patients in the FAS without major protocol violations, was used for supportive analyses.
Primary and secondary efficacy analyses, except for the incidence of endophthalmitis and rescue medications, were performed by applying the last observation carried forward method to impute missing values if at least one post-baseline value was available. Patients with no post-baseline evaluations were considered “failures” (worst-case approach).
The primary efficacy endpoint was analyzed using a two-group large-sample normal approximation test of proportions. The non-inferiority of test treatment vs. the standard therapy was met if the lower bound of the two-sided 95% CI around the treatment difference was >−10%.
A sensitivity analysis was performed on the primary efficacy endpoint by applying the worst-case approach to all dropout patients regardless of the time of study discontinuation. A stratified analysis by centre was also performed using the Cochran–Mantel–Haenszel test.
Descriptive statistics were performed and two-sided 95% CIs of the difference between treatments were computed for the secondary efficacy endpoints by applying a two-group large-sample normal approximation test of proportions.
Safety and tolerability analyses were performed on the Safety Set, defined as all randomized patients who applied at least one dose of study treatment.
Analyses were performed using SAS®, version 9.4 (SAS Institute).
Results
Patients and adherence
Group assignments, loss to follow-up and reasons for withdrawal are summarized in Fig. S2. On 863 patients screened for eligibility, 808 were randomized from August 2018 through November 2018.
The baseline characteristics in the FAS were similar in the two groups as reported in Table 1. Intracameral administration of cefuroxime was carried out in 80% of patients in both treatment arms.
Regarding the compliance, overall adherence was high as 90.89% and 91.86% patients did not miss a dose during the 1st week of treatment in the test arm and control arm, respectively, and 91.26% and 89.74%, respectively, during the 2nd week.
As regards concomitant topical treatments, topical NSAIDs were allowed during the study but were used in only 8% of both the test and the control arms.
Primary outcome
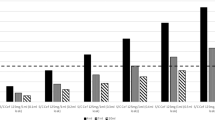
As shown in Table 2 and Fig. 1a, 95.19% of the L-DSP arm and 94.91% of the control arm had no signs of anterior chamber inflammation at day 15. The difference of 0.028 (95% CI: [−0.027; 0.033]) between the two proportions demonstrates the non-inferiority of the L-DSP treatment since the lower bound of the two-sided 95% CI (−0.027) is >−0.10. The outcomes of the sensitivity analysis are consistent with those of the main analysis and are reported in Fig. 2.

a Signs of anterior chamber inflammation; b conjunctival hyperaemia; c TOSS Total Ocular Symptoms Score; d ocular pain and discomfort. a The anterior chamber cell count and flare score were assessed using slit lamp. b Conjunctival hyperaemia was evaluated on a scale ranging from 0 = absence of inflammation to 3 = severe inflammation. c Total Ocular Symptoms Score (TOSS) is the sum of itching/burning, hyperaemia and tearing scores, each symptoms scoring from 0 = none to 3 = severe. d Ocular pain and discomfort were evaluated on a four-point scale, ranging from 0 = absent to 3 = severe.

Last observation carried forward (LOCF) method was used to impute missing values if at least one post-baseline value is available. Dropout patients with only the baseline value are considered as ‘failures’. Worst case: dropout patients independently of the time of dropout are considered as ‘failures’. Stratified: within each country, centres with <10 patients randomized were pooled for the stratified analysis. n = number of patients without signs of anterior chamber inflammation; m = total number of patients valid in the analysis set. Difference between treatment groups is calculated as (levofloxacin + dexamethasone) vs. (tobramycin + dexamethasone). FAS full analysis set, PP per protocol.
Secondary outcomes
No case of endophthalmitis was reported during the study. The outcomes of other secondary endpoints are summarized in Fig. 1. The proportion of patients without signs of inflammation in the anterior chamber at days 4, 8 and 15 is reported in Fig. 1a. The intergroup difference was equal to −0.037 (95% CI: [−0.097; 0.024]) at day 4 and −0.012 (95% CI: [−0.060; 0.036]) at day 8.
The proportion of patients without conjunctival hyperaemia on days 4, 8 and 15 is reported in Fig. 1b. Hyperaemia was mostly mild in both groups and never severe. The intergroup difference was 0.031 (95% CI: [−0.020; 0.083]), −0.030 (95% CI: [−0.072; 0.017]) and −0.015 (95% CI: [−0.046; 0.016]) at days 4, 8 and 15, respectively.
The proportions of patients without ocular symptoms self-assessed through the TOSS at days 4, 8 and 15 were comparable (Fig. 1c). Intergroup differences were equal to 0.034 (95% CI: [−0.025; 0.093]), −0.067 (95% CI: [−0.061; 0.048]) and −0.025 (95% CI: [−0.070; 0.020]) at days 4, 8 and 15, respectively.
Only marginal differences were seen in the proportions of patients complaining of ocular pain and discomfort at days 4, 8 and 15 (Fig. 1d), with intergroup differences equal to −0.007 (95% CI: [−0.046; 0.032]), −0.005 (95% CI: [−0.041; 0.031]) and 0.005 (95% CI: [−0.025; 0.035]) at days 4, 8 and 15, respectively.
Only four patients (0.51%) required rescue therapy for the operated eye, three in the L-DSP arm and one in the Tobradex® arm.
Adverse events
Exposure to study treatment was very similar in the two arms, both in terms of cumulative dose and mean daily dose (Table 3).
The distribution of treatment-emergent adverse events (TEAE) was similar in the two groups except for a slightly higher incidence of headache in the L-DSP arm (Table 3).
Corneal oedema was the most common TEAE and was reported in 3.29% of the L-DSP arm and 4.83% of the Tobradex® arm, but most likely due to the surgical procedure.
In the L-DSP arm, three patients (0.76%) reported at least one severe adverse event. Four patients of the L-DSP arm (1.01%) and two patients of the Tobradex® arm (0.51%) reported serious TEAEs: three patients suffered from a fracture after a fall, two patients (one in each arm) had a myocardial infarction and one patient in the L-DSP arm had a retinal detachment. None of these events were related to treatment. Four patients in the L-DSP arm and three patients in the Tobradex® arm discontinued treatment due to an adverse event. Other observations related to safety (IOP, VA and local tolerability) were very similar in the two treatment arms (Table 3).
Discussion
Cataract surgery causes corneal trauma, ocular inflammation and ocular dryness, which, in turn, result in ocular irritation [27, 28]. Conjunctival hyperaemia, anterior chamber cells and flare and cystoid macular oedema are the most relevant signs of inflammation after cataract surgery [29]. Inflammation is usually self-limiting, but drug therapy is routinely used in clinical practice to shorten resolution time and alleviate ocular discomfort [30], also in relation to the practice patterns in postoperative drug therapy published by the American Society of Cataract Refractive Surgery, the Canadian Ophthalmological Society and the ESCRS [31,32,33]. Corticosteroid eye drops have been used to prevent and treat ocular inflammation for decades, and dexamethasone is the most commonly used [32]. However, the optimal duration of corticosteroid therapy is not defined.
The results of this study have shown that corticosteroid treatment exerts an intense anti-inflammatory activity very quickly both in the anterior chamber and on the ocular surface. Indeed, over 70% of patients in both treatment arms had no inflammatory signs in the anterior chamber, such as 0 cells and 0 flare, after only 3 days. At the end of the 1st week, the percentage of patients without inflammatory signs in the anterior chamber was higher than 85% in both groups, and the other patients (<15%) presented only very modest inflammatory signs. At the end of the 2nd week, inflammatory signs in the anterior chamber (the main outcome) were absent in more than 95% of patients, without significant intergroup differences. Therefore, it is conceivable to consider that in the clear majority of patients undergoing cataract surgery, 7 days of corticosteroid treatment may be sufficient to completely control anterior chamber inflammation.
Consistently, conjunctival hyperaemia was completely controlled after 3 days of therapy in 85.3% of the L-DSP arm and 82.2% of the control arm. After 7 days, conjunctival hyperaemia completely regressed in over 90% of patients in both treatment arms. Therefore, 7 treatment days were also sufficient to completely control the post surgical ocular surface inflammation.
It is important to underline that these anti-inflammatory effects on anterior chamber and ocular surface were not significantly influenced by the concomitant use of topical NSAIDs, which was used by <10% of patients of both groups.
Therefore, to limit the risk of side effects, the prescription of additional therapy with dexamethasone alone after the 1-week levofloxacin/dexamethasone combination may be limited to the very small proportion of patients who still present signs of inflammation of such intensity as to require further treatment. In our study, there was no tapering of the corticosteroid treatment since the recurrence of the inflammatory process at the suspension of the treatment, possible in chronic inflammatory and autoimmune diseases, is unlikely after a surgical trauma once the inflammation has been completely controlled by the therapy. However, more data are needed to understand whether reducing the duration of corticosteroid therapy may have an impact on the incidence of pseudophakic macular oedema (Irvine–Gass syndrome).
Regarding the prophylaxis of infections, ocular infections are largely caused by pathogens colonizing the ocular surface [33], and may occur until the complete closure of the surgical wound. Topical antibiotics are therefore frequently used after the recommended intracameral administration of cefuroxime [11, 34]. The preferred topical antibiotics belong to the aminoglycoside and quinolone families, but the latter is more active against Gram-positive bacteria, frequently responsible for infections after cataract surgery. The duration of topical antibiotic prophylaxis is not standardized. Despite in the ESCRS prophylaxis study of endophthalmitis, topical levofloxacin treatment lasted 6 days [35], topical antibiotics are generally continued in clinical practice for at least 2 weeks. Moreover, to favour patient compliance, the topical antibiotics are often administered in a preformed combination with a corticosteroid. Given the custom of corticosteroid tapering, the antibiotic in fixed associations is also progressively reduced after the initial 2 weeks. These widespread modalities of empirical treatment must be considered irrational since the surgical wound usually heals within 7 days [13, 14]. Therefore, prolonging the administration of a topical antibiotic beyond this period is not reasonable as protracted antibiotic treatment, besides not being useful, could favour the appearance of bacterial resistance, especially if tapering is performed [15,16,17].
To our knowledge, this study is the first to compare two topical antibiotic prophylaxis regimens of different duration: 7 days, justified by the mean surgical wound repair time, vs. 14 days, which represents the average duration implemented in clinical practice. In this study, topical prophylaxis was performed independently of the intracameral administration of cefuroxime, which although recommended by the ESCRS, was not mandatory and was performed in 80% of patients in both groups. During the study, no case of endophthalmitis was observed in both arms. Therefore, the short-term prophylaxis performed in the L-DSP arm was not different from the protracted one implemented in the Tobradex® arm. The results of the study seem to confirm the efficacy of short antibiotic prophylaxis, which, if implemented systematically, would lead to at least halving the amount of antibiotic used in prophylaxis in the most widespread surgical intervention in the world, with undoubted advantages from the point of view of cost containment and reduction of adverse events. Furthermore, this new short-lasting treatment strategy could represent a major step forward in the direction of limiting the prophylactic use of antibiotics and, consequently, the emergence of antibiotic resistance, one of the main public health problems that will have to be effectively addressed in the coming years.
The current study had some limitations, including the single-blind design (however, no alternative was possible because of the switch from a fixed combination to corticosteroid alone in the study arm and the different formulation of the medications: L-DSP is a solution and Tobradex® a suspension, so the blindness could not be ensured), uncomplicated cataract as inclusion criterion, and the number of patients enrolled in relation to the very low incidence of endophthalmitis. In any case, the size of the recruited sample, which is among the highest in the clinical trials available on this specific indication, suggests that with short-term prophylaxis the incidence of endophthalmitis is likely to be <0.2%, and not greater than that expected in a population partially treated with intracameral antibiotic injection [36].
On the other hand, the present study has some strengths: a robust methodology, a conservative sample size for the principal endpoint (one of the highest in literature) and high-quality findings (a very high quote of randomized patients was analyzable and the study was completed in <4 months).
In conclusion, this is the first randomized clinical study showing the validity of short-term antibiotic/corticosteroid treatment in cataract surgery. The present study suggests that a fixed-dose ophthalmic preparation of levofloxacin and dexamethasone may represent a judicious addition to the current armamentarium of drugs available for the treatment/prevention of inflammation and the prevention of infection following cataract surgery. From a pragmatic point of view, this study may also suggest that a follow-up visit should be planned 1 week after surgery as it allows to identify patients that theoretically do not need further medication if have complete resolution of inflammation. This new short-lasting treatment strategy could, therefore, represent both an opportunity to optimize the use of corticosteroids after cataract surgery and a major step forward in the direction of limiting the prophylactic use of antibiotics and, consequently, the emergence of antibiotic resistance.
Summary
What was known before
-
Evidence on medical treatment after cataract surgery is relatively limited to the duration of topical infection prophylaxis (after the recommended intracameral administration of cefuroxime) and of anti-inflammatory treatment. Frequently these treatments are performed with a fixed antibiotic/corticosteroid eye drop combination, but the duration of the administration is not standardized. In the clinical practice, the antibiotic prophylaxis is generally continued for at least 2 weeks, longer than probably necessary, as surgical wound usually repairs within 1 week. Also, the duration of topical corticosteroids (generally 2 or more weeks, often with a dose tapering of the fixed combination) is not supported by clinical evidence, and shortening is potentially convenient as the adverse reactions, including intraocular hypertension, are dose dependent.
What this study adds
-
This study provided for the first time the evidence that 1-week levofloxacin–dexamethasone eye drops were effective as 2-week tobramycin–dexamethasone eye drops for local prophylaxis of ocular infection: no endophthalmitis was observed in either treatment group. Moreover, 1-week of levofloxacin–dexamethasone eye drops were able to abolish ocular inflammation in 85% of patients, showing that for a high percentage of patients it is not necessary to prolong the corticosteroid treatment beyond the 1st week after surgery. Based on the results of this study, which showed that the administration of levofloxacin–dexamethasone in fixed combination for a single week is effective for the prophylaxis of infections and for controlling post surgical inflammation, new strategies could be designed for patients undergoing cataract surgery in order to save misuse of antibiotics and consequently prevent antibiotic resistance as well as to minimize steroidal side effects.
Change history
09 February 2021
A Correction to this paper has been published: https://doi.org/10.1038/s41433-020-0987-9
References
Prokofyeva E, Wegener A, Zrenner E. Cataract prevalence and prevention in Europe: a literature review. Acta Ophthalmol. 2013;91:395–405.
Sharma B, Abell RG, Arora T, Antony T, Vajpayee RB. Techniques of anterior capsulotomy in cataract surgery. Indian J Ophthalmol. 2019;67:450–60.
Cataract surgical rates. Community Eye Health J. 2017;30:88-9.
Eurostat European Commission of the European Community. www.eurostat.ec.europa.eu. Accessed 29 Aug 2018.
European Society of Cataract Refractive Surgery. ESCRS Survey. 2012. http://www.analeyz.com.
Wielders LHP, Schouten JSAG, Winkens B, van den Biggelaar FJHM, Veldhuizen CA, Oliver Findl O, et al. European multicenter trial of the prevention of cystoid macular edema after cataract surgery in nondiabetics: ESCRS PREMED study report 1. J Cataract Refract Surg. 2018;44:429–43.
Kersey JP, Broadway DC. Corticosteroid-induced glaucoma: a review of the literature. Eye. 2006;20:407–16.
Razeghinejad MR, Katz LJ. Steroid-induced iatrogenic glaucoma. Ophthalmic Res. 2012;47:66–80.
Spaeth GL, Monteiro de Barros DS, Fudemberg SJ. Visual loss caused by corticosteroid-induced glaucoma: how to avoid it. Retina. 2009;29:1057–61.
Nallasamy N, Grove KE, Legault GL, Daluvoy MB, Kim T. Hydrogel ocular sealant for clear corneal incisions in cataract surgery. J Cataract Refract Surg. 2017;43:1010–4.
Porela-Tiihonen S, Kokki H, Kaarniranta K, Kokki M. Recovery after cataract surgery. Acta Ophthalmol. 2016;94 Suppl 2 :1–34.
Behndig A, Cochener B, Guell JL, Kodjikian L, Mencucci R, Nuijtis RMMA, et al. Endophthalmitis prophylaxis in cataract surgery: overview of current practice patterns in 9 European countries. J Cataract Refract Surg. 2013;39:1421–31.
Barry P, Cordovés L, Gardner S. ESCRS guidelines for prevention and treatment of endophthalmitis following cataract surgery: data, dilemmas and conclusions. Blackrock, Co. Dublin: European Society of Cataract and Refractive Surgeons, Temple House; 2013.
Gower EW, Lindsley K, Tulenko SE, Nanji AA, Leyngold I, McDonnell PJ. Perioperative antibiotics for prevention of acute endophthalmitis after cataract surgery. Cochrane Database Syst Rev. 2017;2:CD006364.
Barry P. Intracameral antibiotic prophylaxis: American paper mirrors European experience. J Cataract Refract Surg. 2013;39:2–3.
WHO. Worldwide country situation analysis: response to antimicrobial resistance. 2015. https://www.who.int/drugresistance/documents/situationanalysis/en.
Morley GL, Wacogne ID. UK recommendations for combating antimicrobial resistance: a review of “antimicrobial stewardship: systems and processes for effective antimicrobial medicine use” (NICE guideline NG15, 2015) and related guidance. Arch Dis Child Educ Pract Ed. 2017;edpract-2016-311557.
Barlam TF, Cosgrove SE, Abbo LM, MacDougall C, Schuetz AN, Septimus EJ, et al. Implementing an antibiotic stewardship program: guidelines by the Infectious Diseases Society and the Society for Healthcare Epidemiology of America. Clin Infect Dis. 2016;62:e51–77.
Invernizzi A, Marchi S, Aldigeri R, Mastrofilippo V, Viscogliosi F, Soldani A, et al. Objective quantification of anterior chamber inflammation: measuring cells and flare by anterior segment optical coherence tomography. Ophthalmology. 2017;124:1670–7.
Bodaghi B, Weber ME, Arnoux YV, Jaulerry SD, Le Hoang P, Colin J. Comparison of the efficacy and safety of two formulations of diclofenac sodium 0.1% eyedrops in controlling postoperative inflammation after cataract surgery. Eur J Ophthalmol. 2005;15:702–11.
Faraldi F, Papa V, Santoro D, Santoro D, Russo S. Netilmicin/dexamethasone fixed combination in the treatment of conjunctival inflammation. Clin Ophthalmol. 2013;7:1239–44.
Bielory L. Ocular symptom reduction in patients with seasonal allergic rhinitis treated with the intranasal corticosteroid mometasone furoate. Ann Allergy Asthma Immunol. 2008;100:272–9.
Freedman LS. The effect of noncompliance on the power of a clinical trial. Controlled Clin Trials. 1990;11:157–68.
Noble S, Goa KL. Loteprednol etabonate. BioDrugs. 1998;10:329–39.
Stewart R, Horwitz B, Howes J, Novack GD, Hart K. A double-masked, placebo-controlled evaluation of 0.5% loteprednol etabonate in the treatment of post-operative inflammation. J Cataract Refract Surg. 1998;24:1480–9.
A double-masked, placebo-controlled evaluation of 0.5% loteprednol etabonate in the treatment of post-operative inflammation. The loteprendol etabonate postoperative inflammatory study Group 2. Ophthalmology. 1998;105:1780–6.
Li XM, Hu L, Hu J, Wang W. Investigation of dry eye disease and analysis of the pathogenic factors in patients after cataract surgery. Cornea. 2007;26:S16–20.
Rosado-Adames N, Afshari NA. The changing fate of the corneal endothelium in cataract surgery. Curr Opin Ophthalmol. 2012;23:3–6.
Kasetsuwan N, Satitpitakul V, Changul T, Jariyakosol S. Incidence and pattern of dry eye after cataract surgery. PLoS One. 2013;8:e78657.
Dell SJ, Hovanesian JA, Raizman MB, Crandall AS, Doane J, Snyder M, et al. Randomized comparison of postoperative use of hydrogel ocular bandage and collagen corneal shield for wound protection and patient tolerability after cataract surgery. J Cataract Refract Surg. 2011;37:113–21.
DeCroos FC, Afshari NA. Perioperative antibiotics and anti-inflammatory agents in cataract surgery. Curr Opin Ophthalmol. 2008;19:22–6.
Juthani VV, Clearfield E, Church RS. Non-steroidal anti-inflammatory drugs versus corticosteroids for controlling inflammation after uncomplicated cataract surgery. Cochrane Database Syst Rev. 2017;7:CD010516.
Wu CM, Wu AM, Young BK, Wu D, Chen A, Margo CE, et al. An evaluation of cataract surgery clinical practice guidelines. Br J Ophthalmol. 2015;99:401–4.
Ong-Tone L, Bell, Tan YY. Practice patterns of Canadian Ophthalmological Society members in cataract surgery: 2011 survey. Can J Ophthalmol. 2012;47:124–30.
Endophthalmitis Study Group, European Society of Cataract & Refractive Surgeons Prophylaxis of postoperative endophthalmitis following cataract surgery: results of the ESCRS multicenter study and identification of risk factors. J Cataract Refract Surg. 2007;33:978–88.
Shorstein NH, Winthrop KL, Herrinton LJ. Decreased postoperative endophthalmitis rate after institution of intracameral antibiotics in a Northern California eye department. J Cataract Refract Surg. 2013;39:8–14.
Acknowledgements
We would thank OPIS (Desio, Italy), the CRO for the trial, for the fruitful and skilful assistance in managing and analyzing the data.
LEADER-7 Investigators
Pasquale Aragona11, Paolo Arvedi12, Carlo Cagini13, Luigi Caretti14, Gian Maria Cavallini15, Salvatore Cillino16, Innocente Figini17, Livio Marco Franco18, Alberto La Mantia19, Antonio Laborante20, Paolo Lanzetta21, Mattia Marcigaglia22, Cesare Mariotti23, Enrico Martini24, Leonardo Mastropasqua25, Simonetta Morselli26, Franco Passani27, Alfredo Pece28, Grazia Pertile29, Antonino Pioppo30, Cesare Pirondini31, Marcello Prantera32, Antonio Rapisarda33, Mario R. Romano34, Giuseppe Scarpa35, Domenico Schiano-Lomoriello36, Vincenzo Scorcia37, Gianluca Scuderi38, Francesco Semeraro39, Franco Spedale40, Giovanni Staurenghi41, Daniele Tognetto42, Marco Tosi43, Giuseppe Trabucchi44, Fausto Trivella45, Edoardo Villani46, Andrea Vento47, Paolo Vinciguerra48, Jorge L. Alió49, Josè F. Alfonso Sanchez50, Francisco Arnalich Montiel51, Katrin Lorenz52, Irina Panova53, Alena Eremina54, Giorgio Ciprandi55
Funding
NTC Milan, Italy, sponsored the study.
Role of the funding source
The study sponsor had the following role in: study design, data collection, analysis, interpretation of data, writing of the report and decision to submit the paper for publication.
Author information
Authors and Affiliations
Consortia
Corresponding author
Ethics declarations
Conflict of interest
FB reports grants from NTC srl, Allergan, Fidia-SOOFT, Novartis, Sifi; TK reports grants from Allergan, Avedro, Bausch & Lomb, Carl Zeiss, Dompé, Geuder, Hoya, Johnson & Johnson, Med Update, Merck, Novartis/Alcon, Oculentis, Oculus, Presbia, Rayner, Santen, Schwind, Staar, Tear Lab, Thea, Thieme, Zeiss, Ziemer; BM reports grant from NTC srl. All other authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Member of the LEADER-7 Investigators are listed below Acknowledgements.
Rights and permissions
About this article

Cite this article
Bandello, F., Coassin, M., Di Zazzo, A. et al. One week of levofloxacin plus dexamethasone eye drops for cataract surgery: an innovative and rational therapeutic strategy. Eye 34, 2112–2122 (2020). https://doi.org/10.1038/s41433-020-0869-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41433-020-0869-1
This article is cited by
-
Theranostic contact lens based on cellulose nanofibrils/levofloxacin nanocomposite for ocular bacterial infection
Cellulose (2023)
-
Evaluation of the effect of gentamicin in surgical perfusion solution on cataract postoperative endophthalmitis
BMC Ophthalmology (2022)
-
A One-Week Course of Levofloxacin/Dexamethasone Eye Drops: A Review on a New Approach in Managing Patients After Cataract Surgery
Ophthalmology and Therapy (2022)
