
Abstract
In Asian populations, polypoidal choroidal vasculopathy (PCV), a distinct phenotype of neovascular age-related macular degeneration (AMD), is more prevalent than Caucasians. Recently, there has been significant focus on how PCV differs from typical AMD. Although typical AMD and PCV share a variety of mechanisms by which abnormal angiogenic process occurs at the retinochoroidal interface, PCV has different clinical characteristics such as aneurysm-like dilation at the terminal of choroidal neovascular membranes, less frequent drusen and inner choroidal degeneration due to the thickened choroid. Recent studies support an important role for inflammation, angiogenesis molecules and lipid metabolism in the pathogenesis of neovascular AMD. Furthermore, although less attention has been paid to the role of the choroid in AMD, accumulating evidence suggests that the choriocapillaris and choroid also play a pivotal role in drusenogenesis, typical AMD and PCV. This review discusses the basic pathogenic mechanisms of AMD and explores the difference between typical AMD and PCV.
摘要
息肉状脉络膜血管病变 (PCV) 是新生血管性年龄相关性黄斑变性 (AMD) 的一种特殊表型, 好发于亚洲人群, 较高加索人更为常见。近年来研究集中关注于PCV与典型AMD的不同之处。尽管典型AMD和PCV在视网膜脉络膜交界面异常血管生成的过程中具有多种相似的发病机制, 但PCV具有不同的临床特征, 如脉络膜新生血管末端瘤样扩张, 由脉络膜肥厚引起的内层脉络膜变性, 较少见玻璃膜疣等。最新研究提出, 炎症、血管生成因子和脂类代谢均在新生血管性AMD的发生中起到重要作用。此外, 虽然研究较少关注脉络膜在AMD中的作用, 但是大量证据表明脉络膜毛细血管和脉络膜在典型AMD和PCV玻璃膜疣发生中起到关键作用。本综述讨论了AMD的基本发病机制, 探讨了典型AMD和PCV之间的差异。
Similar content being viewed by others


Introduction
Age-related macular degeneration (AMD) is a chronic, progressive disease with two end stages, exudative AMD and geographic atrophy (GA). Exudative AMD is characterized by choroidal neovascularization (CNV), which leads to leakage of fluid, lipids, and blood in the retina. Exudative AMD is more common than GA and an important cause of irreversible vision loss in Asian countries [1]. With an aging population, the size of the affected patient population will inexorably continue to increase. It is estimated that 288 million people will be affected by AMD by 2040, with 113 million people in Asia [1]. The patient’s quality of life (QOL) is diminished by visual impairment due to AMD to the same extent as that of serious systemic diseases, and although current treatments offer a good cost–benefit balance in terms of medical economics, significant improvements in QOL remain a distant prospect. The presence of drusen and retinal pigment epithelial (RPE) abnormalities which are clinical signs of early and intermediate AMD respectively, are associated with an increased risk of progression to advanced AMD.
Importantly, exudative AMD in Asian countries is now categorized into typical AMD and polypoidal choroidal vasculopathy (PCV). When the frequency of exudative AMD classified as typical AMD and PCV was investigated, 35–61% and 33–55% patients diagnosed with AMD were reported to be suffering from typical AMD and PCV respectively [2, 3]. PCV accounts for a much larger proportion of cases in Asia compared to that in Europe and North America, and in recent years, the pathology of PCV has been widely studied in Asian countries using a variety of approaches.
Basic definitions of PCV
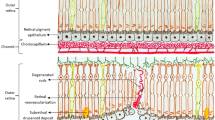
PCV meets the late AMD criteria in the International ARM epidemiological study group and is categorized as a form of neovascular AMD [4]. However it has distinct clinical characteristics from typical AMD (Fig. 1). The definitive diagnosis of PCV is made by indocyanine green (ICG) angiography which reveals a branching vascular network (BVN) with adjoining polypoidal lesion(s) [5]. It is generally agreed that BVNs are the growth of abnormal blood vessels from the choroid and located in Bruch’s membrane, a similar location to the occult CNV observed in typical AMD. Polypoidal lesions show either coil-like or aneurysmal configuration on ICG angiography. Histologically, neovascularization in PCV is characterized by pronounced degenerative processes in choroidal vessels, such as loss of vascular endothelial cells and α-smooth muscle actin positive smooth muscle cell loss [6, 7]. (Fig. 2) Such degenerative changes in the choroid are possibly responsible for the aneurysm-like dilation of the neovascular membrane and common bleeding events observed in PCV patients. Other distinct features include relatively younger age, male predominance, and unilateral presentation.

Multimodal imaging of typical AMD and PCV

H&E staining of surgically excised tissue removed from PCV and typical AMD. PCV (left panel): H&E staining shows dilated vessels, with thickened and hyalinised vessel walls. There is also obstruction of hyalinised vessels. Typical AMD (right panel): H&E staining shows fibrovascular tissues with fibrosis, fibroblast-like cells and small vascular channels. Original magnification: x50
To begin with, the pathogenic mechanisms and processes of typical AMD and PCV are complex and multi-layered, which brings about many key areas of potential research and therapeutics.
Pachychoroid and PCV
It is widely accepted that a substantial proportion of PCV occurs in eyes with a “pachychoroid” configuration and that the choroid is a key contributor to the development of PCV. (Fig. 3) “Pachychoroid” is defined as characteristic anatomical and functional alterations of the choroid including increased choroidal thickness, which may be diffuse or focal, and/or choroidal hyperpermeability on ICG angiography. The most salient feature of the pachychoroid is “pachyvessels” or dilated choroidal veins at the level of Haller’s layer with thinning of the overlying Sattler’s layer and choriocapillaris. Based on multimodal imaging studies, pachychoroid has been postulated to be associated with increased hydrostatic pressure in the choroid causing disruption of Bruch’s membrane, RPE dysfunction, and inner choroidal ischemia [8], and has been considered an underlying cause of a disease spectrum that includes pachychoroid pigment epitheliopathy, central serous chorioretinopathy, and pachychoroid neovasculopathy (type 1 neovascularization associated with shallow irregular RPE detachment with or without polyps). It is likely that pachychoroid is also associated with focal choroidal excavation [9], geographic atrophy (pachychoroid geographic atrophy [10]), peripapillary exudative changes (peripapillary retinoschisis [11] and peripapillary pachychoroid syndrome [12]) and drusen with a peculiar shape (pachydrusen [13]), although there is some debate as to whether pachychoroid is a primary cause of these conditions.

Pachychoroid spectrum diseases. “Pachychoroid” is defined as characteristic anatomical and functional alterations of the choroid, including increased choroidal thickness and/or increased choroidal hyperpermeability on ICG angiography. Pachychoroid is considered responsible for pachychoroid spectrum diseases including pachychoroid pigment epitheliopathy, central serous chorioretinopathy and pachychoroid neovasculopathy. CSR central serous chorioretinopathy, PPE pachychoroid pigment epitheliopathy, PNV pachychoroid neovasculopathy
The recent expansion of our understanding of pachychoroid has raised fundamental questions as to whether PCV differs from typical AMD. Indeed, pachychoroid neovasculopathy includes not only PCV, but also type 1 CNV in typical AMD. Now that PCV and type 1 CNV associated with pachychoroid are understood as pachychoroid-driven disorder, some researchers assume that these 2 conditions are not distinct diseases but simply represent different stages of the same spectrum disorder. However, despite a growing body of literature, it is difficult to diagnose pachychoroid unambiguously due to a lack of expert consensus on the definition. Further studies are needed to reconstruct the classification of neovascular AMD.
Basic mechanisms of typical AMD and PCV
PCV is a multifactorial disease with both genetic and environmental factors contributing to its pathogenesis. Both typical AMD and PCV arise from abnormal blood vessels, resulting in fluid accumulation, recurrent exudative, and hemorrhagic pigment epithelial detachments with associated serous macular detachment.
Genetic factors in typical AMD and PCV
Susceptibility genes associated with PCV are similar to those associated with typical AMD. Fan et al. demonstrated that 34 known AMD loci accounted for up to 36.8% of phenotypic variations for PCV and that there is a high genetic correlation between typical AMD and PCV [14]. Gene loci associated with PCV susceptibility are implicated in complement and immune response, angiogenesis signaling pathways, lipid transport, oxidative stress pathways, and extracellular matrix (ECM), and they are involved in a variety of mechanisms by which abnormal angiogenic processes occur at the retinochoroidal interface.
There is some evidence to suggest there are different genetic factors between typical AMD and PCV. For example, FGD6 is significantly associated with PCV but not with typical AMD [15]. In addition, there are differences in the effect of rare coding variants on AMD susceptibility between European and Asian population. Of note, CETP (D442G) is an East Asian-specific but otherwise uncommon mutation strongly associated with neovascular AMD with an odds ratio of 1.70 [16]. The study also reported three other novel loci (C6orf223 A231A, SLC44A4 D47V, and FGD6 Q257R) associated with neovascular AMD susceptibility in East Asian. Target sequencing of AMD susceptibility in Japanese patients also identified enrichment of low-frequency variants in CETP, C2, and CFB [17], whereas in European populations, there was no significant enrichment of rare variants in these genes. Such differences may represent genuine differences in genetic architecture in AMD between Asians and Europeans.
However, it is widely accepted that there is an overall similarity in the background genetic factors of typical AMD and PCV. Emerging clinical evidence [18, 19] suggest that some of the earliest events in typical AMD start with alterations in the RPE/Bruch’s membrane and the choriocapillaris complex [20, 21]. Subsequent pathways of the pathogenesis of typical AMD and PCV differ, especially with regards to the extent of the involvement of the RPE/Bruch’s membrane and the choriocapillaris.
Age-related changes of RPE in AMD
The RPE plays important roles in retinal homeostasis as follows; (1) provision of nutrients needed for photoreceptor cells; (2) transportation of ions, water, metabolic end products from the subretinal space to the choroid; (3) formation of the blood retinal barrier; (4) phagocytosis of the shed photoreceptor outer segments, and (5) secretion of growth factors. Bruch’s membrane is lamellar extracellular material (ECM) composed of five layers, i.e., inner basal lamina of RPE, outer collagenous zone, elastic zone, inner collagenous zone, and outer basal lamina of choriocapillaris. Bruch’s membrane inner border is well demarcated, whereas the outer limit is ill-defined with expansions of Bruch’s membrane in the choroid forming intercapillary pillars [22]. Progressive age-related changes that characterize early AMD includes thickening of Bruch’s membrane, accumulation of extracellular deposits such as basal laminar deposit and membranous debris. There is an age-related reduction in hyaluronic acid [23] as well as accumulation of proteoglycans [24], collagen [25], elastin [23], lipids [26], and advanced glycation end products [27]. These pathological changes at the level of Bruch’s membrane have been considered to play important roles in AMD. Mechanistically, a vast amount of literature shows that age-related thickening of Bruch’s membrane results in impaired diffusion of oxygen, nutrients, ion, water, and metabolic end products from the RPE and thereby causing RPE dysfunction. Drusen are membranous debris located between the RPE basement membrane and the inner collagenous layer [28] and are considered to be incompletely digested material from RPE [20]. Soft drusen are a clinical hallmark of early AMD and are considered as independent risk factors for late AMD. These drusen are composed of proteins such as complement and β-amyloid, and lipids including unesterified cholesterol and cholesteryl-esters. Importantly, soft drusen are less frequently observed in eyes with PCV compared to those with typical AMD [29, 30] and its role in PCV pathogenesis is less clear.
Age-related changes of the choriocapillaris in AMD
The choroid is a vascularized tissue with poor autoregulatory mechanisms and consists of four layers; (1) outermost suprachoroid lamina, (2) Haller’s layer (large choroidal veins), (3) Sattler’s layer (medium-sized choroidal arterioles and venules), and (4) choriocapillaris (inner sinusoidal capillary network). Choroidal stroma is composed of pigmented cells, fibroblasts, melanocytes, and ECM such as collagen and elastin. The choroid also has multiple functions such as (1) nutrient transport, (2) waste removal, and (3) immune cell tracking [31]. Transport between the choroid and the RPE occurs at the level of choriocapillaris, a thin sheet (7–10 μm) of fenestrated capillaries. There is a close association between the degeneration of the RPE and choriocapillaris. Although less attention has been paid to the role of the choroid in AMD, accumulating evidence suggests that the choriocapillaris and choroid also play a pivotal role in drusenogenesis, typical AMD and PCV [32]. First, there is an age-related thinning of the choriocapillaris and capillary lumen diameters [33]. Although the precise etiology and biochemical pathway is unknown, such changes of the choriocapillaris are considered as a process of normal aging and are present before the onset of clinical manifestation. Histological studies appear to support a role for widened intercapillary stroma, but not sclerotic changes of the choriocapillaries, for these changes [33]. Concomitantly, there is ghost vessels formation due to loss of functional endothelium. Such changes with normal aging are also of clinical significance. In early AMD, drusen are closely related to the choriocapillaries associated with the collecting venules of the choroid [34]. It is postulated that decreased choriocapillary vessel density results in drusen formation due to insufficient clearance of RPE-derived debris. In neovascular AMD, histological studies have demonstrated that there was loss of choriocapillaris adjacent to CNV and large areas that lacked choriocapillaries were reportedly completely covered by RPE [21]. Interestingly, areas with active CNV always had reduced choricapillaries with intact RPE. This contrasts to geographic atrophy, where there are some areas of normal choriocapillaris with atrophied RPE [21]. Furthermore, in typical AMD, CNV is frequently localized to watershed zone as demonstrated by ICG angiography [35]. The watershed zone is considered to be most vulnerable to ischemia in the event of a fall in the perfusion pressure in the vascular bed, and is therefore susceptible to CNV development. The finding that choroidal blood flow is decreased in eyes with an increased risk of CNV in AMD [36] and in eyes with RPE hypertrophy [37] supports this hypothesis. Therefore, the loss of choriocapillaris and reduced choroidal flow are also important factors contributing to exudative AMD [21], although there has been a debate as to whether the choriocapillaris degeneration is a cause or consequences of the inflammatory response that causes the pathogenic changes in the choroidal/RPE ECM.
Contrary to the prevailing view that the ischemia is associated with CNV in typical AMD, abnormal vessels in PCV frequently co-localize with dilated choroidal vessels[38]. Additionally, choroidal vascular hyperpermeability and choroidal thickening are more frequently reported in PCV compared to typical AMD [39]. The most likely mechanism is thickening and increased rigidity of the sclera, probably due to a change in the structural composition of the sclera in the eyes with PCV similar to those with uveal effusion syndrome; that is, dilation of the choroidal vessels could be related to an increased resistance of choroidal venous flow due to impeded venous drainage via vortex veins. Genetic factors are likely to play a role, although the detailed mechanisms are still unknown. Recent investigators postulated that the dilated outer choroidal vessels would mechanically compress the inner choroid and choriocapillaris and might induce ischemic changes [40].
Thus, pathologic changes occurring at RPE/choroidal interface and choriocapillaris play a critical role in the pathogenesis of typical AMD and PCV. However, differing histological and clinical manifestations of PCV from those of typical AMD suggests that there are factors that contribute to the different physical characteristics of the angiogenic process. As discussed in the next chapter, several hypotheses have been postulated; however the main differences in abnormal angiogenic process between typical AMD and PCV are still unknown.
Mechanistic insights into the pathogenesis of typical AMD and PCV
Inflammation - complement and immune response
Complement activation in drusen
It is widely accepted that cellular damage induced by inflammation as part of the complement activation pathway of our immune system contributes to the pathogenesis of typical AMD and PCV. The complement system is a part of the innate immune system, which helps to protect host cells from pathogens, removes debris and modulates immune reactions. There are three major pathways of complement activation, i.e., classical pathway, lectin pathway and alternative pathway. Activation of C3 by a C3 convertase, regardless of how it is activated, results in the generation of anaphylatoxins C3a and C5a, opsonins C3b, iC3b, and C3d, and membrane attack complex (MAC) C5b-9 (Fig. 4) [41]. MAC formation is the final event occurring downstream of complement cascade which induces dysfunction of the target cells by forming membrane-spanning pores [42]. It has been also demonstrated the formation of anaphylatoxins C3a and C5a, resulting from dysregulation of complement pathways, can elicit aberrant activation of macrophages further aggravating the inflammation. Based on histological findings, complement activation in the drusen has been considered to play an important pathogenic role in AMD [20, 41, 43]. A polymorphism in the complement factor H (CFH) gene, a negative regulator of the complement pathway, increases the risk of AMD and PCV by 1.75 and 1.85 fold respectively [14]. Other gene loci in the complement pathways (C2-CFB-SKIV2I, C3, C9, and CFI) have consistently been shown to be associated with AMD risk. In this regard, it is of note that the complement component C3 and other complement pathway proteins are detected in the drusen. Our groups and others found there is an age-related increase in the complement pathway protein C3 [44]. Drusen also trigger further inflammation and C-reactive protein, a CFH-binding protein [45], is also implicated in this process. Drusen accumulation is also enhanced by local inflammation resulting from RPE disorders [20].

Complement pathway. C3 convertase generated either by the classical, lectin, or alternative pathway, cleaves C3 into C3a and C3b, and results in C3b amplification loop. Cascade then proceeds to the activation of a C5 convertase of the classical (C4b2aC3b) or alternative (C3bBbC3b) pathway. Further, C5 convertase initiates the activation of late components of the complement system to form membrane attack complex (MAC). MAC formation is the final event occurring downstream of complement cascade inducing dysfunction of the target cells by forming membrane-spanning pores. Both complement factor H (CFH) and factor I (CFI) promote the cleavage of C3b to its inactive form and act as negative regulators. Genetic studies have identified associations of multiple SNPs associated complement genes including CFH, CFI, CFB, C2, C3, and C9, and AMD susceptibility
Complement activation in the choriocapillaris
While many studies have focused on the impact of complement activation on RPE cells, several studies indicate complement activation first occurs in the choriocapillaris. First, although RPE is exposed to MAC in advanced AMD, activation of MAC first occurs at the level of choriocapillaris in early AMD as demonstrated by a histological study [42]. In addition, the eyes harboring the risk allele of CFH (Y402H) shows increased C-reactive protein in the choroid [46], probably reflecting chronic local inflammation and cellular injury. Moreover, in neovascular AMD, persistent labeling of MAC at the level of the choriocapillaris is reported even after the degeneration of the endothelium was complete. These lines of evidence suggest complement activation occurs in the choriocapillaris at initial stage of AMD. Interestingly, complement activation also triggers innate inflammatory responses in choroidal endothelial cells. Choriocapillaris is the only capillary system in which the endothelial cells express intercellular adhesion molecule-1 (ICAM-1), an important molecule that mediates the infiltration of leukocytes [47]. In vitro, C5a increases the expression of ICAM-1 [48], suggesting complement activation contributes to the recruitment of leukocytes. Further evidence to support the role of complement pathway molecule on the choroid comes from animal experiments. In aged mice, there was deposition of amyloid beta and tau proteins in the choroidal arteries, and both amyloid β and phosphorylated tau were expressed at significantly high levels in CFH knockout mice [49]. Amyloid β and phosphorylated Tau in association with brain blood vessels are known to disrupt endothelial cell function; therefore, it is plausible that the accumulation of these proteins results in the disruption of choroidal vessels, potentially thinning the choriocapillaris and thus disrupts outer retinal perfusion.
Role of immune cells
Among infiltrating leukocytes, the role of macrophages has been most intensively studied. Macrophages accumulate around drusen and remove debris and cholesterol from the Bruch’s membrane in the young choroid. In contrast, macrophages also have opposite destructive effects and are involved in the pathogenesis of CNV [50]. (Fig. 5) We and other groups have demonstrated that macrophages recruited to CNV lesion express vascular endothelial growth factor (VEGF) -A and depletion of monocytes and macrophages results in a reduced CNV size in a laser-induced mouse model [51]. Such diverse effects of macrophages may be carried out by distinct macrophage subpopulations. We speculate that macrophages’ polarization from protective to destructive phenotypes is due to continuous stimulation by molecular “garbage” such as oxidized lipid products whose generation-disposal balance becomes impaired with aging. In contrast to the roles of macrophages, roles of lymphocytes and granulocytes in CNV are less clear [52]. Of note a recent study identified mast cell degranulation in the choroid of all stages of AMD [53]. Mast cells were distributed in the choroidal stroma at the level of Sattler’s and Haller’s layer but were not present at the level of choriocapillaris except for the eyes with geographic atrophy where mast cells were also found at the level of choriocapillaris in regions with complete RPE atrophy. Although further studies are needed, it is interesting to postulate that mast cells would release diverse factors that mediate pro- or anti-inflammatory functions which lead to thinning of the choroid, degradation of vascular membranes and Bruch’s membrane, resulting in RPE degeneration and thinning of the choriocapillaris which contribute to the pathogenesis of CNV.

Diagram showing roles of myeloid cells in AMD. Macrophages accumulate around drusen and remove debris and cholesterol from the Bruch’s membrane in the young choroid. In contrast, macrophages have opposite destructive effects and are involved in the neovascular process. Recruited macrophages replace choroidal capillaries, erode Bruch’s membrane including intercapillary pillars and facilitate CNV growth. Macrophages also produce diverse proangiogenic factors such as vascular endothelial growth factor (VEGF) and inducible nitric oxide synthase (iNOS), further promoting angiogenesis
The innate immune response occurring in the choroid is likely to be associated with the pathogenesis of PCV. As mentioned previously, while the concept that drusen-triggered activation of immune system contributes to typical AMD is widely accepted, there remain several controversies with regards to the role of drusen in PCV. Unfortunately, there are no histological studies investigating the choroid of eyes with PCV; however, immunohistological studies using surgically excised PCV specimen showed CD68-positive macrophages around the hyalinised vessels [7], supporting the role of macrophages in the pathogenesis of PCV. Recent evidence suggests that functional skewing of immune cells towards pro-angiogenic and pro-inflammatory phenotype is a key event happening in exudative AMD. The immune cells are shown to be plastic, and polarized cells have the capacity to redifferente and reprogrammed upon various stimuli. Therefore, future studies on the immune cells, especially focusing on their polarization and signaling pathways would pave a way to a new treatment.
The inflammasome has also emerged as another immune component related to AMD [54]. Previous studies reported that the inflammasome, which is typically synthesized by immune cells could be activated in the RPE and could mediate both protective and destructive signals in AMD [55, 56]. This was followed by numerous studies that linked inflammasome activation in RPE and AMD [57]. However, a recent study pointed out the problems related to the use of non-specific and unreliable commercially available antibodies in these experiments and clearly demonstrated no significant NLRP3 (a pivotal cytosolic innate immune sensor and regulator of inflammasome) expression in RPE cells [58]. Their results suggest that RPE cells do not contain significant amounts of NLRP3 that contribute to AMD pathogenesis and call into question the notion that inflammasome activation is involved in the pathogenesis of AMD. Further molecular analysis and independent verification are still awaited to confirm/refute previous studies regarding the roles of inflammasome activation in AMD.
Angiogenesis and signaling pathways
CNV is stimulated by angiogenic factors, which is triggered by ischemia, hypoxia and inflammation. Reduction in anti-angiogenic factors in the RPE may also contribute to the formation of CNV [21]. Bruch’s membrane represents a physical barrier to the inward growth of choroidal vessels. In PCV, it is generally agreed that BVN is localized above the Bruch’s membrane, although some of them can be below the Bruch’s membrane (in the choriocapillaris and the large choroidal vascular layer) [38, 59]. Therefore, CNV in AMD and abnormal blood vessels in PCV require matrix metalloproteases or serine proteases to digest ECM or basement membrane of the choroid, to penetrate into the Bruch’s membrane. Interestingly, although most of the risk variants for geographic atrophy and neovascular AMD are shared, a variant upstream of MMP9 was found to be exclusively associated with neovascular AMD, but not with GA [60], supporting the idea MMP9 is involved in the angiogenic process in neovascular AMD. The digestion of ECM is also mediated by macrophages at least in part. At the initiation stage of CNV, macrophages are shown to accumulate in areas where Bruch’s membrane is thin. There are three possible roles of choroidal/Bruch’s membrane macrophages in the pathogenesis of CNV, i.e., replacement of choroidal capillaries, erosion of Bruch’s membrane including intercapillary pillars and facilitation of CNV [50]. It has been hypothesized that macrophages infiltrate into the compromised areas of Bruch’s membrane and are responsible for digesting collagen. Recruited macrophages also produce diverse proangiogenic factors such as vascular endothelial growth factor (VEGF) and inducible nitric oxide synthase (iNOS) [61], further promoting angiogenesis.
Recent whole-genome exome sequencing identified a significant association between a rare variant in the FGD6 gene (FGD6 K329R) and PCV, but not typical AMD [15]. The authors hypothesized FGD6 would regulate proangiogenic activity in PCV. An abnormal vascular structure that resembles polyps was observed in six out of the 10 retinas receiving the FGD6 R329 virus injection, but only one out of ten in the retinas receiving viruses expressing FGD6 K329 and none in the GFP controls, suggesting that retinas expressing FGD6 R329 have higher probability to cause distorted blood vessels. Their finding suggests that FGD6 R329 can induce the development of an abnormal vascular network in vivo and that this protein might contribute to the unique abnormal vascular network of PCV. Future research in terms of targeting the gene products of FGD6 R329 could be a potential area to explore for neovascular AMD.
Lipid transport
Genome wide association studies have demonstrated an association between Asian exudative AMD and cholesterol ester transfer protein (CETP) [16, 17]. Interestingly Asian specific CETP D442G mutation is known to impair CETP function and increase serum high-density lipoprotein (HDL) cholesterol levels. In Asian population, higher HDL was an independent risk factor for neovascular AMD including typical AMD and PCV [62]. Similarly, genome-wide association studies conducted in Western countries have shown an association between AMD and hepatic lipase (LIPC), which also affects serum HDL cholesterol levels. Additionally, there is an association between apolipoprotein E (ApoE) and ATP-binding cassette transporter A1 (ABCA1) gene and AMD susceptibility, further supporting a link between lipoprotein and AMD. To date, however, the association between serum lipid and AMD has been inconsistent. In contrast to CETP gene, the HDL-raising allele of the LIPC gene was associated with a reduced risk of AMD [63]. Higher total cholesterol and low-density lipoprotein (LDL) levels were associated with an increased risk of AMD, whereas higher HDL levels reduced the risk of advanced AMD. A recent study using Mendelian randomization supports that HDL cholesterol is a causal risk factor for AMD. Unexpectedly, the study found no causal effect of LDL-cholesterol or triglycerides. They also found variants in the CETP gene locus associated with increased HDL is associated with increased AMD risk although variants in LIPC gene locus that increases HDL associated with the opposite effect [64]. Perhaps the racial differences in the effects of the CETP and LIPC gene loci on HDL levels and subsequently AMD risks could be explored in larger studies.
Lipid and drusen
The presence of cholesterol and apoproteins (Apo B, E, A–I, C–I, and C–III) in drusen and basal linear deposits links AMD with lipoproteins involved in the pathogenesis of atherosclerosis, which also features extracellular lipoprotein deposition and end-stage changes including calcification and neovascularization [65, 66]. The retina requires cholesterol to achieve normal structure and function. Local biosynthesis is not the only source of cholesterol for the retina as cholesterol from the systemic circulation can cross the RPE. RPE controls both cholesterol input and output by means of both passive and active mechanisms. RPE uptakes HDL and LDL via scavenger receptor and LDL receptors from the blood circulation and secrete excess fatty acids and cholesterol, ApoB, E. Cholesterol in Bruch’s membrane lipoproteins is likely to be derived from endogenous synthesis, taken-up plasma lipoproteins and phagocytosed photoreceptor outer segment [66]. However, dysregulation of cholesterol uptake and/or removal, together with age-related change in the elastic layer of Bruch’s membrane lead to the accumulation of lipoproteins in the inner collagenous layer with aging. Lipoproteins subsequently form lesions to trigger inflammation, complement activation and cytotoxicity. Due to decreased permeability of Bruch’s membrane, ApoB, E containing lipoproteins secreted by RPE builds up, and individual lipoproteins fuse over time to produce drusen that are largely composed of lipids including unesterified cholesterol and cholesteryl-esters.
Accumulation of lipids is partly due to retention of lipoproteins as a result of impaired reverse cholesterol transport which leads to drusen formation. The importance of HDL and apo-AI for cholesterol efflux has been demonstrated in many studies in non-ocular tissues such as macrophages. In non-ocular tissues, nascent (pre-β-1) HDL particles bind to ABCA1 and promote lipid efflux from the tissue. Moreover, incubation with apoA-I, the major apolipoprotein component of HDL, increases efflux. In this process, ABCA1 transporter on the surface of macrophages transfers free cholesterol from the cells to ApoA-I, which forms pre-β-1 HDL particles. Thereafter, free cholesterol is esterified by lecithin: cholesterol acetyltransferase, and transported to the liver for excretion into bile acid. The ATP-binding cassette G1 transporter (ABCG1) and scavenger receptor class B type I (SR-BI) also contribute to the efflux of cholesterol from peripheral tissues through large and less dense HDL particles. However, the mechanism by which lipids efflux from the RPE into the choroidal circulation is poorly understood. Similar to macrophages, RPE cells express components of reverse cholesterol transport molecules such as apo E, SR-BI, and ABCA1/G1 and lipids are reportedly transported through RPE for efflux from the cells. HDL is shown to bind to the effluxed lipids from the RPE [67]. Interestingly, the RPE cells efflux cholesterol towards both the subretinal and choroidal compartments using ABCA1/G1 [68]. Regarding oxysterols, RPE cells also likely participate in the clearance of dietary oxidized LDL through CD36 scavenger receptor [69]. There are other mediators of cholesterol efflux such as albumin, plasminogen and exosomes in serum. It is also possible that there is monocyte clearance system in which circulating monocytes invade the Bruch’s membrane and accumulate lipid. It is unknown, however, how the recruited monocytes migrate back into the circulation and how much macrophages contribute to the clearance of subretinal lipid. Importantly, several studies demonstrated that increasing cholesterol efflux capacity by means of pharmacological interventions (e.g., statins [70] and ApoA-I mimetic peptides [71]) may reduce drusen burden. However, recent studies suggest that increasing HDL or ApoA-I concentrations do not necessarily improve cholesterol efflux capacity and that HDL functionality is more important [72]. It is also important to note that different statins may differently impact cholesterol efflux capacity, and some types of statins are known to decrease cholesterol efflux [72]. Further studies are warranted to address whether lipid efflux capacity is associated with AMD risk, and if so, which groups of AMD patients would benefit most from such therapy.
Lipids as signaling molecules and fuel substrate
Most of the recent studies on lipids are directed towards understanding the mechanism underlying lipid accumulation in RPE/Bruch’s membrane. However, lipids may affect AMD not only by modulating cellular debris, but also by modulating phenotypic changes of macrophages. In this regard, it is of note that cholesterol metabolism is one of the most important factors that modulate macrophage polarization. Excess cholesterol accumulated in macrophages is removed through ABC cassette transporter, ABCA1/G1, and a recent study demonstrated that down-regulation of ABCA1 in macrophages results in an accumulation of free cholesterol within senescent macrophages [73]. Subsequently, elevated lipid polarizes macrophages to an alternatively activated phenotype that promotes angiogenesis and accelerates neovascular AMD.
It is also important to note lipids are used as fuel substrate. Traditionally, the retina has been assumed to rely exclusively on glucose for energy, and less characterized are the functions of lipid metabolism in the retina as a pathway to generate ATP. However, a recent study clarified that retina uses fatty acid β-oxidation for energy [74]. VLDL receptors (VLDLR) bind triglyceride-rich chylomicrons and VLDLs, allowing lipoprotein lipase to release long-chain fatty acids. Lipid β-oxidation commonly occurs in the heart and skeletal muscle, where abundant amounts of VLDLR facilitate fatty acid uptake. Genetic ablation of Vldlr in mice makes these animals susceptible to neovascularization. In Vldlr knockout mice, the researchers found retinal lipid uptake and lipid β-oxidation were curtailed. Increased level of circulating fatty acids in Vldlr knockout mice could activate a lipid sensor, free fatty acid receptor 1 (Ffar1). This in turn suppresses the expression of the glucose transporter Glut1, thereby impairing glucose entry into photoreceptors. In consequence, fuel shortage and a reduction in the levels of Krebs cycle intermediate α-xketoglutarate promote neovascularization by stabilizing hypoxia inducible factor-1 and VEGF. This mechanism, although needs to be more characterized, may explain neovascular drive due to the reduced availability of lipids that is independent of drusen and inflammation. If proven, therapy against chylomicrons and VLDLs, on top of essential dietary control of lipid intake, could be investigated as a potential preventive therapy for AMD.
Animal models of AMD
Animal models of early AMD
Currently, animal models that accurately mimic the disease pathologies and progression of typical AMD or PCV are still lacking. Several mouse models for AMD were developed based on genetic risk factors. These genetically modified genes in mouse models include complement related genes such as CFH (CFH knock-out mice [75] and knock-in mice expressing human Y402H variant of CFH [76]), cholesterol-related genes such as ApoE (ApoE-null mice [77], ApoE-Leiden mice, and transgenic mice expressing the human ApoE2, ApoE3, or ApoE4 alleles [78]) and oxidative-stress related genes such as SOD1 and 2 (SOD1/2 knockout mice [79, 80]). Although these mouse models replicate some hallmarks of AMD, such as Bruch’s membrane thickening and drusen-like deposits, CNV is rarely present in these models. There had been no studies that demonstrated the thinning of choroicapillaris in animal models. For example, both ApoE knockout mice and transgenice mice, only when fed with high-fat diet, show Bruch’s membrane thickening [77, 78]. CFH knockout mice show minor changes such as a loss of retinal function and increased C3 deposition that occurs at 2 years of age [75], whereas mice expressing either Y402 or H402 variant of human CFH develop C3b deposits and subretinal macrophage infiltration at 12 to 14 months of age [76]. Some animal models exhibit drusen-like deposits; however, these are located in the subretinal space, but not in the sub-RPE space, and differ from human drusen with regards to the composition. Of note, drusen-like deposits can be related with a mutation in CRB1 gene, which is present in several laboratory mouse strains [81], and may have no relevance with the AMD gene mutations these mice carry. Furthermore, CETP, a crucial cholesterol-related gene in Asian AMD, is absent in mice, highlighting the difficulty in replicating the human cholesterol-metabolism abnormality occurring in human AMD in mice. In summary, these mouse models could be useful to investigate the early events occurring in AMD. More studies however will be required to understand how the findings in mice translate to humans to ultimately understand biological and physiological mechanisms in human AMD.
Animal models of late AMD
Developing a mouse model exhibiting cardinal features of typical AMD or PCV has been challenging, and many studies have employed laser-induced CNV model that partially mimic the pathogenic mechanisms of CNV. There is, however, some experimental support for the role of high temperature requirement factor A1 (HtrA1), one of four known proteases belonging to the broadly conserved family of HtrA proteins, in PCV. HtrA1 is linked to typical AMD/PCV and regulates the transforming growth factor β and matrix metalloproteinases in chronic inflammation. Although there is some debate, AMD/PCV-associated single nucleotide polymorphisms in the promoter of HtrA1 has been shown to be associated with an increased expression level of HtrA1 [82,83,84]. HtrA1 risk allele is associated with increased HtrA1 expression. Recently, HtrA1 transgenic mouse strains have been developed by three independent research groups and are reported to show a large number of phenotypes that resemble the human PCV [85,86,87], i.e., (1) capillary structural abnormalities in the choroid; (2) without any other signs of AMD; and (3) the degradation of choroid vessel walls.
Jones et al. generated a mouse line overexpressing human HtrA1 in mouse RPE [86]. The mice expressed human HtrA1 in the RPE and secreted HtrA1 was also detected in the basal side of RPE, i.e., Bruch’s membrane and in the choroid. On ICG angiography, the investigators detected cardinal features of PCV, namely, numerous hyperfluorescent dots, large polypoidal lesions, and branching vascular network in 59% of the mice they examined. Histologically, the PCV-like lesions in the HtrA1 transgenic mice contained abnormally dilated, thin-walled vessels beneath the RPE. Severe degeneration of the elastic lamina or tunica media of choroidal vessels, as well as fragmentation of the elastic layer in Bruch’s membrane, were observed in the same model. These studies demonstrated that transgenic mice overexpressing human HtrA1 show PCV-like abnormal capillary structures in the choroid. In these mice, the tunica adventitia, which contains a large amount of collagen fibers and the tunica media, which is composed predominantly of smooth muscle, were severely degenerated in choroid arteries. Histological analysis showed that the integrity of the elastic lamina of the Bruch’s membrane was also severely compromised; however, the choroidal vessels were reportedly more severely damaged than that of the Bruch’s membrane.
Vierkotten et al. generated transgenic mice overexpressing mouse HtrA1 in the RPE [87]. The investigators found ultrastructural changes in the elastic layer of Bruch’s membrane of the HtrA1 transgenic mice up to one year. Expression of major components of the ECM as well as ECM proteins involved in elastogenesis were changed; there was a degradation of fibronectin with the generation of fibronectin fragments, together with a reduction of fibulin 5 and tropoelastin.
Nakayama et al. also generated mice ubiquitously overexpressing mouse HtrA1 [85]. Importantly, fluorescein angiography demonstrated leakage in the late phase suggestive of CNV in these mice. Histological study demonstrated subretinal CNV formation originating from the choroid as well as ruptures in the Bruch’s membrane. These various mouse models are useful in conducting future research trials for AMD.
Functional biology of HtrA1
HtrA1 is involved in divergent biological pathways and has also been implicated in several pathologies including cancer, Alzheimer’s disease, arthritis and familial ischemic cerebral small-vessel disease. For example, HtrA1 is downregulated in cancer of diverse origins, and functions to regress solid tumors. Diseases such as the cerebral arteriopathy, autosomal recessive with subcortical infarcts, and leukoencephalopathy (CARASIL) syndrome and the cerebral small vessels disease (CSVD) are caused by mutations or deletions that impair HtrA1 function. In Alzheimer’s disease, HtrA1 is involved in the degradation of tau protein [88] and Apo-E protein [89]. HtrA1 is also upregulated in inflammatory diseases such as osteoarthritis and rheumatoid arthritis.
HtrA1 belongs to the serine peptidase family and is composed of 4 distinct protein domains: an Insulin-like growth factor binding domain (IGF-BD), a Kazal-type motif (KM), a trypsin-like peptidase (proteolytic) domain, and a PDZ domain. Majority of HtrA1 is found in extracellular space, and signal peptide in the N-terminus of the protein is essential for both the expression and the secretion of the HtrA1 protein. Secreted HtrA1 is involved in the homeostasis of the ECM. The remaining fraction localizes to the cytoplasm, microtubules and nucleus.
Degradation of divergent ECM proteins
HtrA1 has been shown to promote degradation of a substantial number of ECM components, such as fibronectin, type III collagen, decorin, aggrecan. ECM proteins. HtrA1 is able to proteolytically degrade apoE4. Protein fragments or aggregates that are produced or degraded by HtrA1 either cause disease or modulate disease. It may also be possible that proteolytic cleaved fragments of extracellular proteins may possess biological activities and HtrA1 is involved in this process [90]. Interestingly, a recent study demonstrated thrombospondin-1 is a substrate for HtrA1, and proteolytically released fragment of thrombospondin-1 promotes tube formation by endothelial cells [91]. Moreover, independent of catalytic activity, secreted HtrA1 inhibits transforming growth factor (TGF)-β/BMP signaling pathways by interacting with their receptors and inhibit Wnt pathway by interacting with β-catenin [92].
Degradation of growth factors
HtrA1 also degrades growth factors such as insulin-like growth factor-binding protein 5 (IGF-BP-5), fibroblast growth factor-8 (FGF8) and TGF-β, [93] thereby functioning as an antagonist. Conversely, macrophage inhibitory factor was also shown to interact with HtrA1 and inhibit the proteolytic activity of HtrA1 [94].
Intracellular HtrA1 and tubulins
Intracellular HtrA1 is localized to microtubules in a PDZ domain-dependent, nocodazole-sensitive manner and promotes polymerization of microtubules. Downregulation of HtrA1 attenuates cell migration [95]. The microtubule-associated protein tau aggregates into intracellular neurofibrillary tangles. HtrA1 has recently been reported to have a non-proteolytic activity that enables it to disentangle Tau neurofibrillary tangles, suggesting that it can regulate the levels and aggregation of Tau. In the RPE, intracellular HtrA1 regulates degradation of tubulin [96]. Using RPE monolayer culture, the investigators also found degradation of tubulin results in impairment of phagocytotic activity of RPE cells [96].
Functions of HtrA1 in the context of CNV
Despite a number of in vitro studies, the functions of HtrA1 in vivo remain largely unknown. A recent study has demonstrated there is a prominent immune complex deposition, complement activation, and infiltration of inflammatory cells in mice showing severe PCV phenotype [97]. A HtrA1 inhibitor was ineffective in treating existing lesions, but anti-inflammatory glucocorticoid was effective in preventing PCV progression. We conclude that PCV initiation is caused by ECM protein-mediated proteolytic degradation due to increased HtrA1 activity while progression is driven by chronic inflammation. It is unclear whether HtrA1 upregulates VEGF expression, with studies showing upregulation in HtrA1 transgenic mice [86] and decreased expression in HtrA1 knockout mice [98] whereas other studies showing no change using HtrA1 transgenic mice [87], suggesting upregulation of VEGF, if any, is not direct consequence of overexpressed HtrA1, but is probably due to secondary events after progression of the disease. A recent study using human induced pluripotent stem cells (iPSCs)-derived RPE cells from patients diagnosed with AMD showed that iPSC-derived RPE cells expressed AMD-related proteins and those from the AMD donors had significantly increased complement and inflammatory factors, which are most exaggerated in HtrA1 lines [99], supporting the hypothesis that HtrA1 is associated with chronic inflammation. Mechanistically, the degradation of the elastic layer is likely caused by elastase activity of HtrA1; [86] however, another study failed to detect any elastase activity of HtrA1 [87]. It is also possible that increased fibronectin fragments, that are degraded by HtrA1, stimulate the release of cytokines and matrix metalloproteases from the RPE [87]. With conflicting evidence of the effects of HtrA1 on VEGF levels and other inflammatory mediators on AMD, it remains to be confirmed by future studies in the pathogenesis of AMD.
Future directions
A pathogenic model of PCV described in this review is summarized in Figs. 6 and 7. Degradation of the choriocapillaris and Bruch’s membrane, which can be initiated by matrix metalloproteases and serine proteases, HtrA1, that leads to thinning of the choriocapillaris and dilated choroidal vessels may be one of the underlying mechanisms responsible for PCV. Further studies are necessary to understand how and why typical AMD and PCV are different. We also need to understand how immune systems in the choroid change with aging and disease. Specific areas that need to be developed to understand the molecular basis of PCV include a comprehensive understanding of the functions of genes associated with PCV risk focusing more on the choroid. Continued work using animal models would provide a more accurate and detailed picture of how inflammation, angiogenic factors and lipid metabolism contribute to PCV pathogenesis. Although HtrA1 transgenic mice do not fully replicate all of the robust phenotypes seen in humans, the HtrA1 transgenic mouse model provides the best opportunity to investigate the pathogenesis of PCV and may have advantages over human samples in terms of genetic simplicity and limited influence of environmental factors.

Pathology of PCV. a Normal retina. b Lipoproteins accumulate in Bruch’s membrane, however, soft drusen are less frequently observed in eyes with PCV compared to those with typical AMD. Although complement pathway molecules are found in drusen in the context of typical AMD, its activation typically takes place in the choriocapillaris when drusen are absent. Activation of complement pathway molecules, and matrix metalloproteases and serine proteases, HtrA1 degrade ECM. Subsequent macrophage mobilization exacerbates inflammation. c Breakdown of Bruch’s membrane takes place before inward progression of abnormal blood vessels occurs. Angiogenesis is driven by macrophage and RPE derived VEGF, iNOS, FDG6, and other cytokines

A proposed pathogenic model of PCV is represented here. Degradation of the choriocapillaris and Bruch’s membrane, which can be initiated by complement activation, leukocyte recruitment and inflammatory mediator lead to thinning of the choriocapillaris and dilated choroidal vessels may be one of the underlying mechanisms responsible for PCV
References
Wong WL, Su X, Li X, Cheung CM, Klein R, Cheng CY, et al. Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: a systematic review and meta-analysis. Lancet Glob Health. 2014;2:e106–116.
Maruko I, Iida T, Saito M, Nagayama D, Saito K. Clinical characteristics of exudative age-related macular degeneration in Japanese patients. Am J Ophthalmol. 2007;144:15–22.
Mori K, Horie-Inoue K, Gehlbach PL, Takita H, Kabasawa S, Kawasaki I, et al. Phenotype and genotype characteristics of age-related macular degeneration in a Japanese population. Ophthalmology. 2010;117:928–38.
Bird AC, Bressler NM, Bressler SB, Chisholm IH, Coscas G, Davis MD, et al. An international classification and grading system for age-related maculopathy and age-related macular degeneration. The International ARM Epidemiological Study Group. Surv Ophthalmol. 1995;39:367–74.
Koh AH, Expert PCVP, Chen LJ, Chen SJ, Chen Y, Giridhar A, et al. Polypoidal choroidal vasculopathy: evidence-based guidelines for clinical diagnosis and treatment. Retina. 2013;33:686–716.
Okubo A, Sameshima M, Uemura A, Kanda S, Ohba N. Clinicopathological correlation of polypoidal choroidal vasculopathy revealed by ultrastructural study. Br J Ophthalmol. 2002;86:1093–8.
Nakashizuka H, Mitsumata M, Okisaka S, Shimada H, Kawamura A, Mori R, et al. Clinicopathologic findings in polypoidal choroidal vasculopathy. Invest Ophthalmol Vis Sci. 2008;49:4729–37.
Gal-Or O, Dansingani KK, Sebrow D, Dolz-Marco R, Freund KB. Inner choroidal flow signal attenuation in pachychoroid disease: optical coherence tomography angiography. Retina. 2018;38:1984–1992.
Chung H, Byeon SH, Freund KB. Focal choroidal excavation and its association with pachychoroid spectrum disorders: a review of the literature and multimodal imaging findings. Retina. 2017;37:199–221.
Takahashi A, Ooto S, Yamashiro K, Tamura H, Oishi A, Miyata M, et al. Pachychoroid geographic atrophy. Ophthalmol Retina. 2018;2:295–305.
Lee JH, Park HY, Baek J, Lee WK. Alterations of the lamina cribrosa are associated with peripapillary retinoschisis in glaucoma and pachychoroid spectrum disease. Ophthalmology. 2016;123:2066–76.
Phasukkijwatana N, Freund KB, Dolz-Marco R, Al-Sheikh M, Keane PA, Egan CA et al. Peripapillary Pachychoroid Syndrome. Retina. 2017; Publish Ahead of Print: 1.2018;38;9:1652-67.
Spaide RF. Disease expression in nonexudative age-related macular degeneration varies with choroidal thickness. Retina. 2018;38:708–16.
Fan Q, Cheung CMG, Chen LJ, Yamashiro K, Ahn J, Laude A, et al. Shared genetic variants for polypoidal choroidal vasculopathy and typical neovascular age-related macular degeneration in East Asians. J Hum Genet. 2017;62:1049–55.
Huang L, Zhang H, Cheng CY, Wen F, Tam PO, Zhao P, et al. A missense variant in FGD6 confers increased risk of polypoidal choroidal vasculopathy. Nat Genet. 2016;48:640–7.
Cheng CY, Yamashiro K, Chen LJ, Ahn J, Huang L, Huang L, et al. New loci and coding variants confer risk for age-related macular degeneration in East Asians. Nat Commun. 2015;6:6063.
Momozawa Y, Akiyama M, Kamatani Y, Arakawa S, Yasuda M, Yoshida S, et al. Low-frequency coding variants in CETP and CFB are associated with susceptibility of exudative age-related macular degeneration in the Japanese population. Hum Mol Genet. 2016;25:5027–34.
Ueta T, Iriyama A, Francis J, Takahashi H, Adachi T, Obata R, et al. Development of typical age-related macular degeneration and polypoidal choroidal vasculopathy in fellow eyes of Japanese patients with exudative age-related macular degeneration. Am J Ophthalmol. 2008;146:96–101.
Nomura Y, Ueta T, Iriyama A, Inoue Y, Obata R, Tamaki Y, et al. Vitreomacular interface in typical exudative age-related macular degeneration and polypoidal choroidal vasculopathy. Ophthalmology. 2011;118:853–9.
Hageman GS, Luthert PJ, Victor Chong NH, Johnson LV, Anderson DH, Mullins RF. An integrated hypothesis that considers drusen as biomarkers of immune-mediated processes at the RPE-Bruch’s membrane interface in aging and age-related macular degeneration. Prog Retin Eye Res. 2001;20:705–32.
Bhutto I, Lutty G. Understanding age-related macular degeneration (AMD): relationships between the photoreceptor/retinal pigment epithelium/Bruch’s membrane/choriocapillaris complex. Mol Aspects Med. 2012;33:295–317.
Korte GE, D’Aversa G. The elastic tissue of Bruch’s membrane. Arch Ophthalmol. 1989;107:1654–8.
Green WR, Enger C. Age-related macular degeneration histopathologic studies. The 1992 Lorenz E. Zimmerman Lecture. Ophthalmology. 1993;100:1519–35.
Pauleikhoff D, Harper CA, Marshall J, Bird AC. Aging changes in Bruch’s membrane. A histochemical and morphologic study. Ophthalmology. 1990;97:171–8.
Marshall GE, Konstas AG, Lee WR. Collagens in ocular tissues. Br J Ophthalmol. 1993;77:515–24.
Holz FG, Sheraidah G, Pauleikhoff D, Bird AC. Analysis of lipid deposits extracted from human macular and peripheral Bruch’s membrane. Arch Ophthalmol. 1994;112:402–6.
Handa JT, Verzijl N, Matsunaga H, Aotaki-Keen A, Lutty GA, te Koppele JM, et al. Increase in the advanced glycation end product pentosidine in Bruch’s membrane with age. Invest Ophthalmol Vis Sci. 1999;40:775–9.
Sarks SH. Ageing and degeneration in the macular region: a clinico-pathological study. Br J Ophthalmol. 1976;60:324–41.
Ueta T, Obata R, Inoue Y, Iriyama A, Takahashi H, Yamaguchi T, et al. Background comparison of typical age-related macular degeneration and polypoidal choroidal vasculopathy in Japanese patients. Ophthalmology. 2009;116:2400–6.
Fujimura S, Ueta T, Takahashi H, Obata R, Smith RT, Yanagi Y. Characteristics of fundus autofluorescence and drusen in the fellow eyes of Japanese patients with exudative age-related macular degeneration. Graefes Arch Clin Exp Ophthalmol. 2013;251:1–9.
Nickla DL, Wallman J. The multifunctional choroid. Prog Retin Eye Res. 2010;29:144–68.
Gelfand BD, Ambati J. A revised hemodynamic theory of age-related macular degeneration. Trends Mol Med. 2016;22:656–70.
Ramrattan RS, van der Schaft TL, Mooy CM, de Bruijn WC, Mulder PG, de Jong PT. Morphometric analysis of Bruch’s membrane, the choriocapillaris, and the choroid in aging. Invest Ophthalmol Vis Sci. 1994;35:2857–64.
Friedman E, Smith TR, Kuwabara T. Senile choroidal vascular patterns and drusen. Arch Ophthalmol. 1963;69:220–30.
Ross RD, Barofsky JM, Cohen G, Baber WB, Palao SW, Gitter KA. Presumed macular choroidal watershed vascular filling, choroidal neovascularization, and systemic vascular disease in patients with age-related macular degeneration. Am J Ophthalmol. 1998;125:71–80.
Grunwald JE, Metelitsina TI, Dupont JC, Ying GS, Maguire MG. Reduced foveolar choroidal blood flow in eyes with increasing AMD severity. Invest Ophthalmol Vis Sci. 2005;46:1033–8.
Xu W, Grunwald JE, Metelitsina TI, DuPont JC, Ying GS, Martin ER, et al. Association of risk factors for choroidal neovascularization in age-related macular degeneration with decreased foveolar choroidal circulation. Am J Ophthalmol. 2010;150:40–47 e42.
Alasil T, Ferrara D, Adhi M, Brewer E, Kraus MF, Baumal CR, et al. En face imaging of the choroid in polypoidal choroidal vasculopathy using swept-source optical coherence tomography. Am J Ophthalmol. 2015;159:634–43.
Dansingani KK, Balaratnasingam C, Naysan J, Freund KB. En face imaging of pachychoroid spectrum disorders with swept-source optical coherence tomography. Retina. 2016;36:499–516.
Ferrara D, Waheed NK, Duker JS. Investigating the choriocapillaris and choroidal vasculature with new optical coherence tomography technologies. Prog Retin Eye Res. 2016;52:130–55.
Anderson DH, Radeke MJ, Gallo NB, Chapin EA, Johnson PT, Curletti CR, et al. The pivotal role of the complement system in aging and age-related macular degeneration: hypothesis re-visited. Prog Retin Eye Res. 2010;29:95–112.
Mullins RF, Schoo DP, Sohn EH, Flamme-Wiese MJ, Workamelahu G, Johnston RM, et al. The membrane attack complex in aging human choriocapillaris: relationship to macular degeneration and choroidal thinning. Am J Pathol. 2014;184:3142–53.
Anderson DH, Mullins RF, Hageman GS, Johnson LV. A role for local inflammation in the formation of drusen in the aging eye. Am J Ophthalmol. 2002;134:411–31.
Yuda K, Takahashi H, Inoue T, Ueta T, Iriyama A, Kadonosono K, et al. Adrenomedullin inhibits choroidal neovascularization via CCL2 in the retinal pigment epithelium. Am J Pathol. 2012;181:1464–72.
Bhutto IA, Baba T, Merges C, Juriasinghani V, McLeod DS, Lutty GA. C-reactive protein and complement factor H in aged human eyes and eyes with age-related macular degeneration. Br J Ophthalmol. 2011;95:1323–30.
Johnson PT, Betts KE, Radeke MJ, Hageman GS, Anderson DH, Johnson LV. Individuals homozygous for the age-related macular degeneration risk-conferring variant of complement factor H have elevated levels of CRP in the choroid. Proc Natl Acad Sci USA. 2006;103:17456–61.
McLeod DS, Lefer DJ, Merges C, Lutty GA. Enhanced expression of intracellular adhesion molecule-1 and P-selectin in the diabetic human retina and choroid. Am J Pathol. 1995;147:642–53.
Skeie JM, Fingert JH, Russell SR, Stone EM, Mullins RF. Complement component C5a activates ICAM-1 expression on human choroidal endothelial cells. Invest Ophthalmol Vis Sci. 2010;51:5336–42.
Aboelnour A, Kam JH, Elnasharty MA, Sayed-Ahmed A, Jeffery G. Amyloid beta deposition and phosphorylated tau accumulation are key features in aged choroidal vessels in the complement factor H knock out model of retinal degeneration. Exp Eye Res. 2016;147:138–43.
Cherepanoff S, McMenamin P, Gillies MC, Kettle E, Sarks SH. Bruch’s membrane and choroidal macrophages in early and advanced age-related macular degeneration. Br J Ophthalmol. 2010;94:918–25.
Tan X, Fujiu K, Manabe I, Nishida J, Yamagishi R, Nagai R, et al. Choroidal neovascularization is inhibited via an intraocular decrease of inflammatory cells in mice lacking complement component C3. Sci Rep. 2015;5:15702.
Tan X, Fujiu K, Manabe I, Nishida J, Yamagishi R, Terashima Y, et al. Choroidal neovascularization is inhibited in splenic-denervated or splenectomized mice with a concomitant decrease in intraocular macrophage. PLoS One. 2016;11:e0160985.
Bhutto IA, McLeod DS, Jing T, Sunness JS, Seddon JM, Lutty GA. Increased choroidal mast cells and their degranulation in age-related macular degeneration. Br J Ophthalmol. 2016;100:720–6.
Datta S, Cano M, Ebrahimi K, Wang L, Handa JT. The impact of oxidative stress and inflammation on RPE degeneration in non-neovascular AMD. Prog Retin Eye Res. 2017;60:201–18.
Doyle SL, Campbell M, Ozaki E, Salomon RG, Mori A, Kenna PF, et al. NLRP3 has a protective role in age-related macular degeneration through the induction of IL-18 by drusen components. Nat Med. 2012;18:791–8.
Tarallo V, Hirano Y, Gelfand BD, Dridi S, Kerur N, Kim Y, et al. DICER1 loss and Alu RNA induce age-related macular degeneration via the NLRP3 inflammasome and MyD88. Cell. 2012;149:847–59.
Kerur N, Fukuda S, Banerjee D, Kim Y, Fu D, Apicella I, et al. cGAS drives noncanonical-inflammasome activation in age-related macular degeneration. Nat Med. 2018;24:50–61.
Kosmidou C, Efstathiou NE, Hoang MV, Notomi S, Konstantinou EK, Hirano M, et al. Issues with the specificity of immunological reagents for NLRP3: implications for age-related macular degeneration. Sci Rep. 2018;8:461.
Hong YJ, Miura M, Makita S, Ju MJ, Lee BH, Iwasaki T, et al. Noninvasive investigation of deep vascular pathologies of exudative macular diseases by high-penetration optical coherence angiography. Invest Ophthalmol Vis Sci. 2013;54:3621–31.
Fritsche LG, Igl W, Bailey JN, Grassmann F, Sengupta S, Bragg-Gresham JL, et al. A large genome-wide association study of age-related macular degeneration highlights contributions of rare and common variants. Nat Genet. 2016;48:134–43.
Ando A, Yang A, Mori K, Yamada H, Yamada E, Takahashi K, et al. Nitric oxide is proangiogenic in the retina and choroid. J Cell Physiol. 2002;191:116–24.
Cheung CM, Laude A, Yeo I, Tan SP, Fan Q, Mathur R, et al. Systemic, ocular and genetic risk factors for age-related macular degeneration and polypoidal choroidal vasculopathy in Singaporeans. Sci Rep. 2017;7:41386.
Reynolds R, Rosner B, Seddon JM. Serum lipid biomarkers and hepatic lipase gene associations with age-related macular degeneration. Ophthalmology. 2010;117:1989–95.
Burgess S, Davey Smith G. Mendelian randomization implicates high-density lipoprotein cholesterol-associated mechanisms in etiology of age-related macular degeneration. Ophthalmology. 2017;124:1165–74.
Curcio CA, Johnson M, Huang JD, Rudolf M. Aging, age-related macular degeneration, and the response-to-retention of apolipoprotein B-containing lipoproteins. Prog Retin Eye Res. 2009;28:393–422.
Pikuleva IA, Curcio CA. Cholesterol in the retina: the best is yet to come. Prog Retin Eye Res. 2014;41:64–89.
Ishida BY, Duncan KG, Bailey KR, Kane JP, Schwartz DM. High density lipoprotein mediated lipid efflux from retinal pigment epithelial cells in culture. Br J Ophthalmol. 2006;90:616–20.
Storti F, Raphael G, Griesser V, Klee K, Drawnel F, Willburger C, et al. Regulated efflux of photoreceptor outer segment-derived cholesterol by human RPE cells. Exp Eye Res. 2017;165:65–77.
Picard E, Houssier M, Bujold K, Sapieha P, Lubell W, Dorfman A, et al. CD36 plays an important role in the clearance of oxLDL and associated age-dependent sub-retinal deposits. Aging. 2010;2:981–9.
Vavvas DG, Daniels AB, Kapsala ZG, Goldfarb JW, Ganotakis E, Loewenstein JI, et al. Regression of some high-risk features of age-related macular degeneration (AMD) in patients receiving intensive statin treatment. EBioMedicine. 2016;5:198–203.
Rudolf M, Mir Mohi Sefat A, Miura Y, Tura A, Raasch W, Ranjbar M, et al. ApoA-I mimetic peptide 4F reduces age-related lipid deposition in murine bruch’s membrane and causes its structural remodeling. Curr Eye Res. 2018;43:135–46.
Talbot CPJ, Plat J, Ritsch A, Mensink RP. Determinants of cholesterol efflux capacity in humans. Prog Lipid Res. 2018;69:21–32.
Sene A, Khan AA, Cox D, Nakamura R, Santeford A, Kim BM, et al. Impaired cholesterol efflux in senescent macrophages promotes age-related macular degeneration. Cell Metab. 2013;17:549–61.
Joyal JS, Sun Y, Gantner ML, Shao Z, Evans LP, Saba N, et al. Retinal lipid and glucose metabolism dictates angiogenesis through the lipid sensor Ffar1. Nat Med. 2016;22:439–45.
Coffey PJ, Gias C, McDermott CJ, Lundh P, Pickering MC, Sethi C, et al. Complement factor H deficiency in aged mice causes retinal abnormalities and visual dysfunction. Proc Natl Acad Sci USA. 2007;104:16651–6.
Ufret-Vincenty RL, Aredo B, Liu X, McMahon A, Chen PW, Sun H, et al. Transgenic mice expressing variants of complement factor H develop AMD-like retinal findings. Invest Ophthalmol Vis Sci. 2010;51:5878–87.
Dithmar S, Curcio CA, Le NA, Brown S, Grossniklaus HE. Ultrastructural changes in Bruch’s membrane of apolipoprotein E-deficient mice. Invest Ophthalmol Vis Sci. 2000;41:2035–42.
Malek G, Johnson LV, Mace BE, Saloupis P, Schmechel DE, Rickman DW, et al. Apolipoprotein E allele-dependent pathogenesis: a model for age-related retinal degeneration. Proc Natl Acad Sci USA. 2005;102:11900–5.
Imamura Y, Noda S, Hashizume K, Shinoda K, Yamaguchi M, Uchiyama S, et al. Drusen, choroidal neovascularization, and retinal pigment epithelium dysfunction in SOD1-deficient mice: a model of age-related macular degeneration. Proc Natl Acad Sci USA. 2006;103:11282–7.
Justilien V, Pang JJ, Renganathan K, Zhan X, Crabb JW, Kim SR, et al. SOD2 knockdown mouse model of early AMD. Invest Ophthalmol Vis Sci. 2007;48:4407–20.
Mattapallil MJ, Wawrousek EF, Chan CC, Zhao H, Roychoudhury J, Ferguson TA, et al. The Rd8 mutation of the Crb1 gene is present in vendor lines of C57BL/6N mice and embryonic stem cells, and confounds ocular induced mutant phenotypes. Invest Ophthalmol Vis Sci. 2012;53:2921–7.
Iejima D, Nakayama M, Iwata T. HTRA1 overexpression induces the exudative form of age-related macular degeneration. J Stem Cells. 2015;10:193–203.
Ng TK, Liang XY, Lai TY, Ma L, Tam PO, Wang JX, et al. HTRA1 promoter variant differentiates polypoidal choroidal vasculopathy from exudative age-related macular degeneration. Sci Rep. 2016;6:28639.
Liao S-MM, Zheng W, Zhu J, Lewis CA, Delgado O, Crowley MA, et al. Specific correlation between the major chromosome 10q26 haplotype conferring risk for age-related macular degeneration and the expression ofHTRA1. Mol Vis. 2017;23:318–33.
Nakayama M, Iejima D, Akahori M, Kamei J, Goto A, Iwata T. Overexpression of HtrA1 and exposure to mainstream cigarette smoke leads to choroidal neovascularization and subretinal deposits in aged mice. Invest Ophthalmol Vis Sci. 2014;55:6514–23.
Jones A, Kumar S, Zhang N, Tong Z, Yang JH, Watt C, et al. Increased expression of multifunctional serine protease, HTRA1, in retinal pigment epithelium induces polypoidal choroidal vasculopathy in mice. Proc Natl Acad Sci USA. 2011;108:14578–83.
Vierkotten S, Muether PS, Fauser S. Overexpression of HTRA1 leads to ultrastructural changes in the elastic layer of Bruch’s membrane via cleavage of extracellular matrix components. PLoS ONE. 2011;6:e22959.
Poepsel S, Sprengel A, Sacca B, Kaschani F, Kaiser M, Gatsogiannis C, et al. Determinants of amyloid fibril degradation by the PDZ protease HTRA1. Nat Chem Biol. 2015;11:862–9.
Chu Q, Diedrich JK, Vaughan JM, Donaldson CJ, Nunn MF, Lee KF, et al. HtrA1 proteolysis of ApoE in vitro is allele selective. J Am Chem Soc. 2016;138:9473–8.
Munoz SS, Li H, Ruberu K, Chu Q, Saghatelian A, Ooi L, et al. The serine protease HtrA1 contributes to the formation of an extracellular 25-kDa apolipoprotein E fragment that stimulates neuritogenesis. J Biol Chem. 2018;293:4071–84.
Chen CY, Melo E, Jakob P, Friedlein A, Elsasser B, Goettig P. et al. N-Terminomics identifies HtrA1 cleavage of thrombospondin-1 with generation of a proangiogenic fragment in the polarized retinal pigment epithelial cell model of age-related macular degeneration. Matrix Biol. 2018;70:84–101.
Globus O, Evron T, Caspi M, Siman-Tov R, Rosin-Arbesfeld R. High-temperature requirement A1 (Htra1) - a novel regulator of canonical Wnt signaling. Sci Rep. 2017;7:17995.
Kim GY, Kim HY, Kim HT, Moon JM, Kim CH, Kang S, et al. HtrA1 is a novel antagonist controlling fibroblast growth factor (FGF) signaling via cleavage of FGF8. Mol Cell Biol. 2012;32:4482–92.
Fex Svenningsen A, Loring S, Sorensen AL, Huynh HUB, Hjaeresen S, Martin N, et al. Macrophage migration inhibitory factor (MIF) modulates trophic signaling through interaction with serine protease HTRA1. Cell Mol Life Sci. 2017;74:4561–72.
Chien J, Ota T, Aletti G, Shridhar R, Boccellino M, Quagliuolo L, et al. Serine protease HtrA1 associates with microtubules and inhibits cell migration. Mol Cell Biol. 2009;29:4177–87.
Melo E, Oertle P, Trepp C, Meistermann H, Burgoyne T, Sborgi L, et al. HtrA1 mediated intracellular effects on tubulin using a polarized RPE disease model. EBioMedicine. 2018;27:258–74.
Kumar S, Nakashizuka H, Jones A, Lambert A, Zhao X, Shen M, et al. Proteolytic degradation and inflammation play critical roles in polypoidal choroidal vasculopathy. Am J Pathol. 2017;187:2841–57.
Zhang L, Lim SL, Du H, Zhang M, Kozak I, Hannum G, et al. High temperature requirement factor A1 (HTRA1) gene regulates angiogenesis through transforming growth factor-beta family member growth differentiation factor 6. J Biol Chem. 2012;287:1520–6.
Saini JS, Corneo B, Miller JD, Kiehl TR, Wang Q, Boles NC, et al. Nicotinamide ameliorates disease phenotypes in a human iPSC model of age-related macular degeneration. Cell Stem Cell. 2017;20:635–47 e637.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Yanagi, Y., Foo, V.H.X. & Yoshida, A. Asian age-related macular degeneration: from basic science research perspective. Eye 33, 34–49 (2019). https://doi.org/10.1038/s41433-018-0225-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41433-018-0225-x
This article is cited by
-
The hypothetical molecular mechanism of the ethnic variations in the manifestation of age-related macular degeneration; focuses on the functions of the most significant susceptibility genes
Graefe's Archive for Clinical and Experimental Ophthalmology (2024)
-
Contribution of common and rare variants to Asian neovascular age-related macular degeneration subtypes
Nature Communications (2023)
-
Hyaluronidase-1-mediated glycocalyx impairment underlies endothelial abnormalities in polypoidal choroidal vasculopathy
BMC Biology (2022)
-
Pigmentary abnormality without significant drusen as a risk factor for late age-related macular degeneration
Scientific Reports (2022)
-
Effects of cataract surgery in Japanese patients with neovascular age-related macular degeneration
Graefe's Archive for Clinical and Experimental Ophthalmology (2021)
