
Abstract
Objectives
USH2A encodes for usherin, a basement membrane protein in the inner ear and retina. USH2A can cause retinitis pigmentosa (RP) with or without hearing loss. The aim of this study was to detect USH2A mutations in a Chinese cohort of 75 small RP families and 10 Usher syndrome families.
Methods
We performed a direct Sanger sequencing analysis of the USH2A gene to identify mutations for this cohort.
Results
We identified a total of eight mutations in four of the 75 small RP families (5.3%) and two mutations in one of the 10 Usher families (10%); all families were detected to have compound heterozygous mutations. In families with nonsyndromic RP, we identified the compound heterozygous mutations p.Pro4818Leuand p.Leu2395Hisfs*19 in family No. 19114, p.Arg4493His and p.His1677Glnfs*15 in family No.19162, c.8559-2A > G and p.Arg1549* in family No.19123 and p.Ser5060Pro and p.Arg34Leufs*41 in family No.19178. We also identified the heterozygous mutations p.Arg3719His and p.Cys934Trp in family No.19124, which was the Usher syndrome family. These mutations were predicted to be harmful by SIFT, PROVEAN, Mutation Taster or PolyPhen-2.
Conclusions
Our results revealed six novel mutations in the USH2A gene in a Chinese population, which is beneficial for the clinical use of genetic testing of USH2A in patients with autosomal-recessive or sporadic RP and Usher syndrome.
Similar content being viewed by others

Introduction
Retinitis pigmentosa (RP, OMIM 226800) is one of the most common incurable eye diseases [1, 2] and has clinical and genetic heterogeneity [3, 4]. The worldwide prevalence of RP is 1:3,000 to 1:7,000 [5, 6]. Due to defects of the photoreceptor rods, the typical clinical symptom of RP is peripheral visual field loss, then gradual central vision loss, which may eventually cause blindness [5, 6]. RP can be inherited as autosomal dominant (30–40%), autosomal recessive (50–60%), or X-linked (5–15%) [5, 6]. So far, 87 genes have been identified as responsible for RP (https://sph.uth.edu/retnet/sum-dis.htm), including 27 genes for autosomal dominant RP, 57 genes for autosomal recessive RP, and three genes for X-linked RP. Most patients have nonsyndromic RP, while others suffer from associated syndromes. The most common syndromic RP is Usher syndrome, which accompanies RP with hearing loss in autosomal recessive inheritance, with a prevalence of approximately 1:20,000 [7]. Thirteen genes have been identified that may cause Usher syndrome (https://sph.uth.edu/retnet/sum-dis.htm).
The USH2A gene is located in chromosome 1 with 72 exons [8]. USH2A causes 10–15% of recessive RP cases and 30–40% of Usher syndrome type 2 cases. USH2A was first mapped in chr1q in Usher families by Lewis [9] and Kimberling in 1990 [10] and the latter identified USH2A as the candidate gene of Usher syndrome type 2in 1995 for the first time [11]. In 2000, Rivolta et al. identified for the first time missense mutations in the USH2A gene with recessive RP without hearing loss [12], suggesting that mutations in USH2A can cause Usher syndrome or RP only. In this study, we sequenced 75 RP and 10 Usher small families by direct Sanger sequencing of the exons of USH2A. We identified six novel mutations in the USH2A gene causing nonsyndromic RP or Usher syndrome in a Chinese population; this finding provides a theoretical basis for follow-up research and treatment of this disease.
Methods
Patient recruitment and ethics statement
This study was approved by the institutional review boards of Beijing Tongren Hospital and Sichuan Provincial People’s Hospital. All experiments were performed in accordance with relevant guidelines and regulations, including any relevant details. All of the participants received and signed the informed consent form. Seventy-five RP and 10 Usher small families were recruited from 2008 to 2012 from the clinicsoftheEye Center of Beijing Tongren Hospital, Capital Medical University.
The diagnosis of nonsyndromic RP was made in all individuals on the basis of an ophthalmologic examination, including visual testing, a slit-lamp examination, ophthalmoscopy, fundus photography, and electroretinogram (ERG). Patients with Usher syndrome reported hearing loss, which was tested clinically and verified by audiometry when required. Clinical data on each patient were included in this analysis. Venous blood (5 ml) was collected from each participant for DNA extraction with a GentraPuregene Blood DNA kit (China) according to the Gentra® Puregene® Handbook.
Mutation screening
Mutation screening was performed by using the direct Sanger sequencing analysis. The coding region (exons 2–72) of USH2A was amplified by polymerase chain reaction (PCR) in these 75 families. The primers for the PCR amplification were previously reported 13. The coding regions (exons 2–72), including the intron–exon boundary of USH2A, were amplified by polymerase chain reaction (PCR). The amplification of target DNA by the PCR produces copies which may contain errors. To decrease the PCR/sequencing errors, we used pfu DNA polymerase to amplify the regions (PrimeSTAR® HS DNA Polymerase with GC Buffer, KAKARA). High GC buffer was used when amplifying the high GC regions. We used forward and reverse primers to sequence them with the BigDye Terminator v3.1 Cycle Sequencing Kit (ABI Applied Biosystems) according to the manufacturer’s instructions and sequenced by ABI 3730 sequencer. Variant-filtering was based on public and in-house SNP databases, including dbSNP137, 1000Genome project, and ExAC Browser (Beta) Exome Aggregation Consortium data, as well as our internal database. We kept nonsynonymous and splicing variants with MAF < 0.1% for further analysis. The likely inheritance pattern of the genes, the functional impact, and the clinical relevance were analyzed to obtain possible mutations. The functional impact of each variant was predicted by SIFT and PROVEAN tools (http://sift.jcvi.org/), Mutation Taster(http://www.mutationtaster.org/) and PolyPhen-2(http://genetics.bwh.harvard.edu/pph2/PolyPhen-2). For each patient, a deleterious or damaged variant that was observed by at least one prediction tool was considered a mutation. The potential pathogenicities of the filtered variants were then interpreted according to the existing and proposed guidelines from the Standards and Guidelines for the Interpretation of Sequence Variants, to establish a diagnosis of the clinician made the final determination of the relationship of the reported variant(s) to the patient’s phenotype.
Results
Clinical characteristics of patients
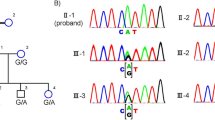
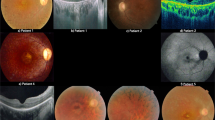
The clinical information of the five patients is shown in Table 1. The pedigrees and fundus photographs of the five patients identified with haveUSH2A mutations are shown in the left and middle panels of Fig. 1. Patient No.19114 had suffered from night blindness since the age of 30; fundus photography displayed fundus pigmentation, and electroretinography (ERG) displayed no amplitude reactionPatient No.19162 had night blindness in both eyes since age 13; fundus photography displayed retinal pigment epithelial (RPE) cell-layer atrophy and vascular narrows, especially in the peripheral retina. There was no ERG reaction. Patient No. 19123 had night blindness since childhood; fundus photography showed a blue-greyperipheral retina, pigmentation, and vision loss. No. 19178 had night blindness since junior high school and was subsequently diagnosed with RP. Fundus photography displayed a concave-shaped reflective area in the central macula and black pigmentation in the posterior pole of the fundus. There was no ERG reaction. Patient No. 19124 had progressively declining vision, with night blindness and hearing abnormalities since age 42. Fundus photography displayed peripheral retinal turbidity and pigmentation resembled the appearance of bone cells. ERG displayed no amplitude reaction, and the patient was diagnosed with Usher-2 syndrome (Table 1).

Pedigree, fundus pictures and chromatograms of detected USH2A mutations. a Pedigree, fundus pictures and chromatograms of detected USH2A mutations in 19114. b Pedigree, fundus pictures and chromatograms of detected USH2A mutations in 19162. c Pedigree, fundus pictures and chromatograms of detected USH2A mutations in 19123. d Pedigree, fundus pictures and chromatograms of detected USH2A mutations in 19178. e Pedigree, fundus pictures and chromatograms of detected USH2A mutations in 19124
USH2A mutation identification
All the variants detected in this study are listed in the Supplementary data. Totally, 1574 variants were detected. After variant-calling and data-filtering, we identified 10 compound mutations in five patients (Table 2), which contained nonsynonymous substitution, splicing, stopgain, frameshift deletion, and frameshift insertion. Four mutations were reported and six were novel. Patient No.19114 had nonsynonymous mutation p.Pro4818Leu (c.14453 C > T) [13] in exon 66 and the frameshift deletion p.Leu2395Hisfs*19 (c. 7183 _7193del) mutation in exon 38 (Table 1 and Table 2). His normal parents carried only one of these two mutations each. Patient No. 019162 had frameshift deletion p.His1677Glnfs*15 (c.5027_5043del) in exon 25 and a missense mutation p.Arg4493His (c.13478 G > A) in exon 63. Patient No. 19123 had a splicing mutant in exon 44 (c. 8559-2 A > G) and a stopgain mutation in exon 22 atArg1549* (c.4645 C > T) [8]. His normal brother only carried the splicing mutation. Patient No.19178 had frameshift insertion mutation p.Arg34Leufs*41(c.100_101insT) [14] in exon 2 and a missense mutation p.Ser5060Pro (c.15178 T > C) in exon70. His brother and sister also suffered from this disease, with the same mutations. The Usher-2 patient, No.19124, carried two missense mutations: p.Cys934Trp (c.2802 T > G) in exon13 and p.Arg3719His (c.11156 G > A) in exon57. Her parents carried one of the heterozygous mutations each, and her daughter wasnormal, carrying neither of these mutations.
Analysis of harmful USH2A mutations by prediction
To determine if the 10 changes identified in the families were pathogenic or not, we performed several prediction analyses. First, we examined the location of the 10 changes along the long usherin isoform and identified most of them located within functional domains (Table 2). Second, we aligned USH2A sequences from different species for each of the changes, all of which were evolutionarily conserved (Fig. 2). Third, based on damage prediction by SIFT, PROVEAN, Mutation Taster, and PolyPhen-2, all of these changes can be predicted as harmful by at least one predictor (Table 2). Thus, the results suggested that the 10 mutations might cause the clinical phenotypes of the patients with RP or Usher syndrome type 2 cases.

Amino acid mutations loci conservative in 8 species. Orthologous alignments of the detected mutations in this study suggest their evolutionarily conservative feature
Discussion
Mutations in the USH2A gene are responsible for the majority of USH2 cases [15, 16]. In addition to typical USH2, a certain mutant allele of the USH2A gene was also found to cause nonsyndromic RP with lessor no hearing defects [12]. USH2A mutations were estimated to underlie approximately7% of all RP cases in North America [17]. A report on Spanish patients showed that mutations in USH2A with autosomal recessive RP had high prevalence and phenotypic variations [18]. However, there is no report focusing on USH2A mutations in large samples of nonsyndromic RP small families in a Chinese population. In this study, we identified five pairs of compound heterozygous mutations in the USH2A gene from 75 nonsyndromic RP patients (5.3%) and 10 Usher patients (10%) in a Chinese population by a direct Sanger sequencing analysis.
USH2A is a large gene with 72 exons, encoding the protein usherin with 5202 amino acids. In mammalian photoreceptors, usherin is localized to a spatially restricted membrane microdomain at the apical inner segment recess that wraps around the connecting cilia, corresponding to the periciliary ridge complex described in amphibian photoreceptors [19]. The targeted defect of the USH2A gene in mice leads to progressive photoreceptor degeneration and moderate but non-progressive hearing impairment, mimicking the visual and hearing deficits in patients with USH2A mutations, suggesting its essential role for the long-term maintenance of retinal photoreceptors and the development of cochlear hair cells [19]. In this study, most of the 10 mutations might be responsible for nonsyndromic RP or USH2 because these changes are located in the functional domains and are predicted to be harmful for the normal function of the protein (Table 2).
Unexpectedly, in the comparison of the recorded mutations of USH2A in nonsyndromic RP to USH2 in the Human Gene Mutation Database (HGMD,http://www.hgmd.cf.ac.uk/ac/index.php), there was no obviously different distribution of these mutations between the two kinds of retinal diseases; the mutations are rather scattered, located in the whole protein. The mutations we detected in this study were located in exons 2, 7, 13, 22, 25, 38, 44, 60, 63, and 70; no more than two mutations were located in the same exon. Using the TMHMM2.0 software prediction for the usherin protein’s subcellular location, we found that nine of the 10 mutations could be located in the extracellular space (Fig. 3, subcellular location). Only one mutation(p.Ser5060Pro) is located in the transmembrane region. The usherin protein has a two-membrane structure domain in both the N and C terminals (Fig. 3, subcellular location). The extracellular space of the usherin protein may cooperate with other USHER proteins in the hair bundle of auditory sensory cells, and in the photoreceptor cells [20]. During the differentiation of the hair bundle, the extracellular space region of the usherin protein may be involved in the formation of a type of stereocilia side link located at the tip and the base of the stereocilia, contributing to the junction between the inner and outer segments, and at the synaptic region in the photoreceptor cells [20]. The defect in the usherin protein from congenital mutations may lead to the disorder of the connecting cilium in the photoreceptors, causing vision loss.

Predicted cellular distribution of the detected mutations.USH2A gene encodes a transmembrane protein. Mutation p.Ser5060Prois predicted in the membrane-bound protein, other mutations are distributed in the exocytoplasmic
So far in the HGMD, most of the mutations have been identified in USH2 patients, while <10 mutations in nonsyndromic RP have been identified. In this study, we identified eight compound mutations in the USH2A gene from 75 patients with nonsyndromic RP and two compound mutations in 10 USHER patients. Six of the mutations we detected are novel. Aside from the severe mutations such as splicing, stopgain, and inframe mutations, the missense mutations are also harmful by prediction. Our results provide valuable information not only for precise clinical gene testing for USH2A in Chinese populations with nonsyndromic RP and USH2, but also for future studies performed to understand the molecular mechanism of USH2A in human disease.
Summary
What was known before
-
The compound heterozygous mutations p. P4818L in family No. 019114 have been identified.
-
The compound heterozygous mutationsc.8559-2A > G and p.R1549X in family No. 019123 have been identified.
-
The compound heterozygous mutations p.R34fs in family No. 019178 have been identified.
What this study adds
-
We identified the compound heterozygous mutations p.2395_2398del in family No. 019114.
-
We identified the compound heterozygous p.R4493H and p.1676_1681del in family No. 019162.
-
We identified the compound heterozygous p.S5060P in family No. 019178.
-
We also identified the heterozygous mutations p. R3719H and p.C934W in family No. 019124, which was the Usher syndrome family.
References
Mitamura Y, et al. Diagnostic imaging in patients with retinitis pigmentosa. J Med Invest. 2012;59:1–11.
Humphries P, Kenna P, Farrar GJ. On the molecular genetics of retinitis pigmentosa. Science. 1992;256:804–8.
Krawczynski MR, Pecold K. [Genetic heterogeneity of retinitis pigmentosa]. Klin Ocz. 1994;96:24–9.
Parmeggiani, F et al. Clinical and rehabilitative management of retinitis pigmentosa: up-to-date. Curr Genomics. 2011;12:250–9.
Hartong DT, Berson EL, Dryja TP. Retinitis pigmentosa. Lancet. 2006;368:1795–809.
Ferrari S, et al. Retinitis pigmentosa: genes and disease mechanisms. Curr Genom. 2011;12:238–49.
Rosenberg T, Haim M, Hauch AM, Parving A. The prevalence of Usher syndrome and other retinal dystrophy-hearing impairment associations. Clin Genet. 1997;51:314–21.
Baux D, et al. Molecular and in silico analyses of the full-length isoform of usherin identify new pathogenic alleles in Usher type II patients. Hum Mutat. 2007;28:781–9.
Lewis RA, Otterud B, Stauffer D, Lalouel JM, Leppert M. Mapping recessive ophthalmic diseases: linkage of the locus for Usher syndrome type II to a DNA marker on chromosome 1q. Genomics. 1990;7:250–6.
Kimberling WJ, et al. Localization of Usher syndrome type II to chromosome 1q. Genomics. 1990;7:245–9.
Kimberling WJ, et al. Gene mapping of Usher syndrome type IIa: localization of the gene to a 2.1-cM segment on chromosome 1q41. Am J Hum Genet. 1995;56:216–23.
Rivolta C, Sweklo EA, Berson EL, Dryja TP. Missense mutation in the USH2A gene: association with recessive retinitis pigmentosa without hearing loss. Am J Hum Genet. 2000;66:1975–8.
Aller E, et al. Identification of 14 novel mutations in the long isoform of USH2A in Spanish patients with Usher syndrome type II. J Med Genet. 2006;43:e55.
Dai H, et al. Identification of five novel mutations in the long isoform of the USH2A gene in Chinese families with Usher syndrome type II. Mol Vis. 2008;14:2067–75.
Eudy JD, et al. Mutation of a gene encoding a protein with extracellular matrix motifs in Usher syndrome type IIa. Science. 1998;280:1753–7.
Pennings RJ, et al. USH2A mutation analysis in 70 Dutch families with Usher syndrome type II. Hum Mutat. 2004;24:185.
Seyedahmadi BJ, Rivolta C, Keene JA, Berson EL, Dryja TP. Comprehensive screening of the USH2A gene in Usher syndrome type II and nonsyndromic recessive retinitis pigmentosa. Exp Eye Res. 2004;79:167–73.
Bernal S, et al. Mutations in USH2A in Spanish patients with autosomal recessive retinitis pigmentosa: high prevalence and phenotypic variation. J Med Genet. 2003;40:e8.
Liu X, et al. Usherin is required for maintenance of retinal photoreceptors and normal development of cochlear hair cells. Proc Natl Acad Sci USA. 2007;104:4413–8.
Bonnet C, El-Amraoui A. Usher syndrome (sensorineural deafness and retinitis pigmentosa): pathogenesis, molecular diagnosis and therapeutic approaches. Curr Opin Neurol. 2012;25:42–9.
Acknowledgements
We would like to thank all the RP patients and their families for participating in this study.
Funding
This research project was supported by: National key research and development program (2016YFC0905200 (ZY)), the National Natural Science Foundation of China (81170883 (ZY), 81430008 (Z.Y.), 81300802 (LH), 81670895 (LH), 81271048 (JY); the Department of Science and Technology of Sichuan Province, China (2014SZ0169, 2015SZ0052 (Z.Y.), 2015JQO057 (LH), 2016HH0072 (LH), 17QNJJ0062 (LH), 2015SZ0060 (YL) and 2013JY0195 (LH).
Author contribution
ZY designed the study. YL and ZY recruited the participants. LH, YM, JY, FL and ZY performed the genotyping. LH and ZY performed the statistical analysis. YM wrote the initial draft, with edits from LH. All authors critically revised and gave final approval of this manuscript.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Huang, L., Mao, Y., Yang, J. et al. Mutation screening of the USH2A gene in retinitis pigmentosa and USHER patients in a Han Chinese population. Eye 32, 1608–1614 (2018). https://doi.org/10.1038/s41433-018-0130-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41433-018-0130-3
This article is cited by
-
USH2A gene variants cause Keratoconus and Usher syndrome phenotypes in Pakistani families
BMC Ophthalmology (2021)
