
Abstract
An increasing number of European research projects return, or plan to return, individual genomic research results (IRR) to participants. While data access is a data subject’s right under the General Data Protection Regulation (GDPR), and many legal and ethical guidelines allow or require participants to receive personal data generated in research, the practice of returning results is not straightforward and raises several practical and ethical issues. Existing guidelines focusing on return of IRR are mostly project-specific, only discuss which results to return, or were developed outside Europe. To address this gap, we analysed existing normative documents identified online using inductive content analysis. We used this analysis to develop a checklist of steps to assist European researchers considering whether to return IRR to participants. We then sought feedback on the checklist from an interdisciplinary panel of European experts (clinicians, clinical researchers, population-based researchers, biobank managers, ethicists, lawyers and policy makers) to refine the checklist. The checklist outlines seven major components researchers should consider when determining whether, and how, to return results to adult research participants: 1) Decide which results to return; 2) Develop a plan for return of results; 3) Obtain participant informed consent; 4) Collect and analyse data; 5) Confirm results; 6) Disclose research results; 7) Follow-up and monitor. Our checklist provides a clear outline of the steps European researchers can follow to develop ethical and sustainable result return pathways within their own research projects. Further legal analysis is required to ensure this checklist complies with relevant domestic laws.
Similar content being viewed by others

Introduction
Increasing numbers of European research projects return, or plan to return, individual genomic research results (IRR), (i.e., findings from clinical research or population-based studies that relate to a single individual) to participants [1,2,3,4,5]. IRR might be study-specific results (SSR) relating to the project’s overarching research question(s), secondary findings (SF; actively sought variants associated with conditions or traits unrelated to the research question), or unsolicited findings (UF; incidentally identified disease-causing variants unrelated to the research question)Footnote 1 [6]. Most projects returning results focus on health-related and actionable IRR (those that can lead to surveillance, prevention, or treatment) as this is considered an ethical priority [7].
The return of IRR is, in principle, supported by diverse stakeholders [8,9,10]. A recent systematic review of 221 empirical articles, encompassing 118,874 individuals across 20 countries (the majority from the USA) identified interest in receiving IRR was high across research participants, patients, and publics, ranging from 47.6–100% [9]. Health professionals, researchers, and institutional review board members are generally more cautious about returning results than participants, patients, and publics were about receiving them [9, 10]. All stakeholders prioritised RoR that could lead to surveillance, prevention and/or treatment. Professionals raised concerns, including difficulties obtaining informed consent, lack of time and resources, possible overdiagnosis, clinical follow-up, and potential for psychological harm [10,11,12,13,14,15,16,17].
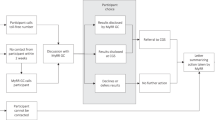
Stakeholders’ views toward return of results (RoR) [8,9,10] reflect those within the European legal framework, which is underpinned by three principles (the participant’s right of access, the participant’s right to know and right not to know, and the researcher’s duty of care), articulated in four legally binding instruments (Fig. 1) [18,19,20,21,22]. In the European Economic Area (EEA), research participants have a right of access to their health data upon request [18,19,20] and, in most EEA countries, also rights to know and not know any information collected about their health [21, 22]. In some EEA countries, researchers have a duty of care to offer participants any information of relevance to their current or future health or quality of life [21, 22] and preferences must be ascertained at study outset [23]. There may be domestic legislation in place creating further obligations; e.g., Italy’s genetic data privacy law offers participants choices about RoR, such as an opt-out (right not to know), whereas Spain’s biomedical research law requires researchers to override a participant’s wish not to know when serious harm to the participant or their relatives can be avoided [24]. Article 89 of the General Data Protection Regulation (GDPR) makes it possible to derogate from the participants’ right of access in EU or member state law where personal data is processed for scientific research purposes, if the right of access is likely to render impossible or seriously impair the achievement of the specific scientific research purposes, and derogation from the right of access is necessary for the fulfilment of those scientific research purposes. In such case, appropriate safeguards, including technical and organizational measures, must be in place.

The three principles underpinning the European framework regarding return of results (the participant’s right of access, the participant’s right to know and right not to know, and the researcher’s duty of care) are articulated here drawing on the four relevant legally binding instruments.
Despite long standing ethical debate [23, 25], there is now broader acceptance that, in addition to the law, there are many ethical and pragmatic reasons to return clinically actionable IRR. While it is generally agreed that participants have ethical, and in several countries legal, rights to receive personal research data, and that researchers may have an obligation to return it, depending on the nature of the relationship established with data contributors [26], the practice of RoR raises several practical issues.
Returning IRR requires researchers to consider the scope of findings to be returned, the strength of evidence for clinical utility, how to organize the informed consent process [27, 28], gather the necessary expertise and resources to interpret variants and return results [29], and establish logistical infrastructure to support the return process [30], and potential follow-up [31]. Recent guidelines provide an overview of steps needed to plan for, and organise, the RoR process [32,33,34]. However, most of these are project-specific [32], focus on which results to return [33] rather than how to organize the RoR process, or were not developed specifically for the European context [34]. The European Society of Human Genetics (ESHG) recommendations for the clinical use of whole genome sequencing suggest caution in the return of UF [35]. These guidelines were not designed to apply to research settings and are now nearly a decade old in a field where technology and genomic data generation are progressing rapidly. Recent ESHG guidance does not recommend active searching for SF in a clinical setting [36].
In 2015, experts participating in the European COST Action “Citizen’s Health through public-private Initiatives: Public health, Market and Ethical perspectives” (CHIP ME) IS1303 [37] network, of which I.B.L and D.F.V were members, recommended development of European harmonized, equitable, scientifically sound, and socially robust guidelines for return of genetic/genomic IRR [38]. Such guidelines would support European researchers and/or biobanks planning and managing RoR, irrespective of differences at micro (research laboratory/group), meso (university/institute/biobank), and macro (region/country) levels. It was argued guidelines should be applicable to both disease-focused and population-based research. Others too have suggested that although it will not always be possible for international collaborations to overcome discrepancies by harmonising their RoR policies and tools, collaborations may want to design RoR processes that still allow for decisions to be made locally [24].
To address this need for practical guidance, and growing interest in RoR, an analysis of normative documents and expert consensus were used to develop a checklist of steps to assist researchers and/or biobanks in Europe considering returning IRR to adult participants to: a) decide whether RoR is appropriate, feasible, and sustainable for their project, b) develop a RoR plan, and c) implement a RoR pathway.
Methodology
The initial drafted checklist was based on existing normative documents that provided guidance on returning IRR. To identify these, a Google search was conducted in October 2020 using the following terms: [(guidance OR guideline OR policy OR recommendation) AND (return OR feedback OR individual OR genetic OR research results OR incidental findings)]. Documents underwent full-text review and were included if they were a) regional, national, or international guidelines providing practical information about steps for RoR; b) publicly available and published in English in the preceding 10 years (based on technological advancements made in this time); and c) for clinical research and/or population/biobank research. They did not need to discuss genetic/genomic research specifically nor be academic publications. Documents were excluded if they only discussed the pros and cons of feedback, only provided a list of genetic variants to return, focused on single aspects of RoR (e.g., feedback to relatives, children, deceased), or were specific to research areas (e.g., epigenetics, psychiatry, imaging) or disease (e.g., cancer).
Documents were read by a member of the research team (I.B.L.) and text that provided information about the steps that needed to be taken or aspects that require consideration to return IRR was extracted into a spreadsheet. Inductive content analysis was used to analyse the data [39]. Data were categorised into broad content categories corresponding to stages within the research process (e.g., planning of research project, informed consent, etc.). Each point within the extracted text was coded (e.g., participants should be offered the option to receive results). Coding of all points was performed by two researchers independently (D.F.V. and I.B.L.). Similar codes were grouped together (D.F.V.) to form subcategories within the broad content categories (e.g., ‘whether results will be returned’ under the ‘develop a plan’ category), then checked and refined (N.H.). All the broad categories and subcategories formed the basis for the first draft of the checklist. As the documents included pertained to RoR in adults, rather than minors, we limited the checklist to RoR in adult research populations.
To gauge the checklist’s relevance to the European context, we sought feedback from a panel of European interdisciplinary experts in RoR-related fields across both clinical research and population-based settings in a seven-hour digital workshop over three days in June 2021. Expert panel members (EPs) were recruited from professional networks of the study team (D.F.V., N.H., I.B.L.), including the COST Action CHIP ME and European Society of Human Genetics, and projects known by the study team to have experience of either returning or planning to return IRR. EPs (n = 27) included: clinicians, clinical researchers, population-based researchers, biobank managers, bioethicists, lawyers and policy makers. EPs watched presentations about several population-based research initiatives that are returning results (HUNT 4, FinHealth P5, Estonia Biobank, 100,000 Genome project), the legal frameworks pertaining to RoR (H.B.B.), and stakeholder perspectives (D.F.V.). The draft checklist was presented and feedback was obtained from EPs using structured discussion facilitated by I.B.L., D.F.V. and N.H. A range of views were presented in the workshop; we report herein the majority consensus (>75% agreement). Discussions were recorded, transcribed, coded and categorised akin to the normative documents. New categories or subcategories arising and agreed upon in the discussion were added to the checklist; none were excluded (I.B.L., D.F.V. and N.H.).
Results
Of the 34 normative documents identified, six met our inclusion criteria, two of which were from Europe/UK (Table 1). The checklist provides an overview of the procedural steps European researchers and/or biobanks should consider to enable RoR to consenting adult research participants, from conception of a project to its full realization, including the production and disclosure of clinically actionable IRR. The checklist applies to both population-based research and clinical research projects. Below we describe the seven steps of the checklist (Table 2), which are intended to guide researchers considering RoR as they 1) decide which results to return; 2) develop a plan for return of results; 3) obtain participant informed consent; 4) collect and analyse data; 5) confirm results; 6) disclose research results; and 7) follow-up and monitor participants and processes.
1. Decide which results to return
Consider the nature of IRR
In addition to the legal requirements for RoR in particular jurisdictions (Fig. 1), decisions regarding which IRR to return to participants should include whether the finding is an SSR, which may depend on whether the study is clinical research or population-based, its potential health or clinical significance [33, 40, 41], its medical actionability [40, 41], and the associated condition [42]. Guidelines suggest clinical experts assist with these determinations [33]. Researchers should consider the potential benefits and harms associated with RoR [41, 43], including participant’s best interests [41], and the potential impact on the participant, including the impact of uncertainty, and the presence/absence of and ability to access an intervention [42]. Countries signatory to the Oviedo Convention’s Additional Protocol on Biomedical Research, have a legal duty of care, which includes returning research information relevant to the current or future health or quality of life, but leaves the interpretation of “care” to the evaluation of researchers and research ethics committees [23, 43]. The findings’ clinical actionability, and analytical and scientific validity should be considered (e.g., positive predictive value, false positive rates) [40, 41, 43], along with limitations on test validity, analysis quality and laboratory accreditation [43] and whether the result will be validated in a clinical laboratory.
Resources should be consulted to support decisions about returning UF (e.g., scientific literature [33], online resources (e.g., ClinVar) [44], internationally accepted guidelines for variant interpretation, (e.g., the American College of Genetics and Genomics; ACMG) [45]). Either a predetermined list or protocol [32], (e.g., the ACMG gene list) [34], or returning exploratory results on a case-by-case basis may be appropriate [42]. Researchers should consider involving clinical expertise in such decisions [23, 32, 43].
Consider context of result return
Researchers should consider the RoR context [22], including study population vulnerability and access to appropriate healthcare [41]. Participant age may also be relevant; EPs mentioned examples where research participants, minors at the time of (parental) consent, reach the age of majority. Participant needs, preferences and values should be incorporated into RoR decision-making [43]. Researchers should follow country-specific good practice guidelines for UF (if they exist) [41] and EPs suggested researchers investigate country-specific funding options/resources and relevant laws to support RoR.
The Australian National Health and Medical Research Council (NHMRC) specifies there is no obligation to assess or return findings beyond research scope/study completion [40].
Consider the practicalities of RoR
If IRR will be returned, researchers must develop a clear disclosure pathway, subject to participant informed consent [40, 41], and determine how to return results in a reasonable timeframe based on when findings will be identified [41]. Consideration should be given to privacy and confidentiality issues relating to data handling, such as the impact of linking identifiers to the samples, collection of excess sample, and family member involvement [42]. Non-European guidelines suggest return decisions regarding both SSR and UF should assess the benefit of return against availability of resources, feasibility and sustainability, including staff training for RoR [41,42,43]. EPs urged researchers to consider identifying appropriate healthcare professionals (e.g., specialist physicians, genetic counsellors), involving them in protocol development, and clarifying the precise role of primary care or specialist healthcare professionals (where relevant) in the disclosure pathway [22]. Logistical requirements requiring consideration include whether researchers (or relevant healthcare teams) can recontact participants [41], whether future analysis is feasible and if so, availability of infrastructure for data storage and reanalysis [40].
Researchers should assess whether RoR will compromise the research aims [46]. One non-European guideline suggests IRR should be offered at study completion if feasible and study integrity is not compromised [42].
2. Develop a plan for return of results
Most guidelines stressed the importance of developing an RoR plan when designing the study [40,41,42,43]. This plan should cover a range of elements (Table 2) including clear descriptions of how (i.e., the intended techniques) [40], and the types of results likely to be generated, including SSR, UF and SF [33, 40,41,42]. EPs emphasised the importance of distinguishing between these types of results and drawing clear distinctions between research and clinical results, as highlighted in the NHMRC National Statement [40]. EPs suggested researchers consider whether separate protocols are needed for return of SSR versus SF/UF. The plan should describe whether, and which types of results will be reported [33, 40, 41, 43]. The rationale for return, which results will be returned at which time points, who will be responsible for making such judgments, return processes for both SSR and UF, and the research team’s responsibilities after result disclosure [46] should be provided [40, 43, 46].
RoR plans should describe how participants’ informed consent and RoR preferences will be sought, and how researchers intend to respond to RoR requests [43], although in the EEA, this is regulated in law. The plan should include whether findings will be validated in a clinical laboratory, the processes for this, whether additional resources or expertise are required [40, 41], how this expertise will be sourced (if not already contained within the research team), and any distinction between analytical and clinical validity [40].
The plan should describe the budget, infrastructure and resources required [43]; both EPs and a US document emphasised ensuring adequate resources to support RoR [43]. They suggest the RoR plan should be described in project funding proposals and that funding bodies incorporate RoR funding into the project budget [43], although EPs recognised this may be challenging. EPs stressed the importance of mentioning whether approval from Data Access Committees will be needed, although they cannot request additional requirements above those imposed by law. The plan should mention any biobanks associated with the study and their role in the RoR process [40], as well as relevant institutional policies [43].
Researchers should seek approval for their project, including the RoR plan, from a Research Ethics Committee (REC) [40, 42, 43, 46], and factor in time for REC changes, which may alter the RoR plan. To ensure sufficient funds for RoR activities, applications for funding should occur prior to seeking REC approval; in some countries, approved funding of projects is a pre-condition for REC approval.
3. Obtain participant informed consent to return of results
When designing the consent process, it is important to ensure the information provided is clear and accessible [41]. Here we focus specifically only on the aspects to be covered in the informed consent (IC) process relating to RoR, not all points required by GDPR. It should discuss the identified through the research (including UF/SF) [33, 41, 46], the likelihood of these occurring [40, 41], which results may be returned to participants and the rationale for return [33, 41,42,43].
Participants should be informed of the potential risks of receiving results [43], including psychological risks and overdiagnosis, any unanticipated risks [42], and insurance implications. Limitations of identified results should be highlighted [41] as should the fact that variant pathogenicity may be reassessed over time. EPs suggested mentioning the potential benefits to participants’ and blood relatives’ health.
Participants should be informed whether they can choose which types of results they wish to receive [41, 42], including not receiving results (i.e., the right not to know as highlighted by EPs and which may be enshrined in national law in most EEA countries) [41, 43, 46]. Researchers should discuss whether participants will be able to reassess their RoR choices [40]. Participants’ decision about RoR should be respected [40, 46]; researchers should have a plan, which may include consulting their relevant REC and domestic law, if serious actionable results are identified in a participant who chose not to receive them [46].
Researchers should describe how results will be returned [33, 40,41,42,43, 46] including whether they will be communicated to participants’ healthcare providers and placed in their medical records [43], and the timeframe and conditions under which results may be returned [40, 43]. They should discuss who will communicate results [40] and the potential implications of the findings [41, 42], such as access to genetic counselling [40, 46], the potential for interventions or follow-up [40, 41], and who will pay for these if healthcare is not free at the point of need [41]. Distinctions between research and clinical care should be made clear to avoid therapeutic misconception [41, 46], as should implications of results for relatives [40] and whether results will be shared with relatives in general or in case of the participant’s death [43].
4. Collect and analyse data
Only one guideline addressed data collection and analysis considerations, which was specifically for UF [33]. This European document suggests researchers should use the tests required to answer the research question but that research teams include healthcare professionals with expertise in handling clinically significant genomic findings [33]. The US Presidential Commission states that researchers may get approval but are not obligated to search for SF [46]. Practically, this might only be possible if the research is conducted in a clinical setting, as seen in the 100,000 Genomes Project [47].
5. Confirm results
Researchers should consider only returning results once they have been validated in a clinical laboratory and the utility of the result established [40]. EPs suggested clinical validation should be done by an accredited laboratory using evidence-based standards (e.g., ACMG) [33, 40, 41, 45]. EPs highlighted the importance of multidisciplinary teams to ensure expert laboratory and clinical input and reporting consistency. One document suggests it may be necessary to involve the participant when confirming analytical validity, (e.g., to collect a second sample for result validation) [41].
6. Disclose research results
Researchers should determine who is responsible for, and will be actively involved in RoR, and how and when this will occur. Guidelines suggest returning IRR should ideally be the responsibility of either an appropriate clinical service or the participant’s clinician in discussion with the research team [40]. EPs stressed it is important to ensure the person providing results is appropriately qualified and certified to country or institution standards [33, 41]. Participants should be informed about the meaning and implications of RoR for themselves and family members [40, 43].
The pathway for returning IRR should be clear and situation-specific [41, 42] and results should be provided in a timely manner [33]. Researchers should consider how results will be returned: in person, by telephone or video conference, via online platform, by a confidential letter, or by a combination of these strategies. If REC approval is obtained to return results directly to participants, researchers should consider engaging with emerging technologies, such as IT-based solutions, to best tailor the information to participants’ needs and preferences [33, 43]. One guideline suggests participants should always be provided with a written summary of the results, regardless of other forms of communication used [43].
7. Follow-up and monitor
Several guidelines state researchers should consider how RoR will allow participants to access clinical follow-up and inform them about this option where appropriate [41, 43]. One guideline, which limits its guidance to UF, suggests researchers should ally with medical specialists to allow clinical follow-up and offer to help participants with this process [33]. EPs suggested researchers should develop a pathway for participants to obtain a clinical referral when needed. One guideline suggests researchers should monitor the effects of communicating UF and evaluate the management of UF policy within the research project [33].
Discussion
We used a mixed methods approach, drawing on existing guidelines and expert perspectives, to develop a practical checklist for researchers and/or biobanks considering RoR to participants. The European legal framework for returning results to research participants includes four legally binding instruments [18,19,20,21,22]. Although they constitute a minimum threshold that must be met to fulfil legal obligations in pan-European projects, these instruments are not ratified in all European countries. As article 89 of the GDPR shows, derogations and additional requirements to these instruments may be given in domestic legislation creating further obligations. Such diversity in international, regional and national laws/policies raises challenges for research combining datasets across multiple jurisdictions [24]. While the law pertaining to RoR may be the same in both population-based research and clinical research contexts, we acknowledge the ethical obligation to return results may vary [27].
When using this checklist, we suggest researchers consider several overarching aspects. First, experiences from previous projects show returning results requires extensive resources to establish appropriate and sustainable infrastructure, obtain the necessary approvals, and assign staff to RoR [48]; these may be difficult to fully assess and/or acquire before the RoR process begins [7, 31]. The suggestion for results to be returned by an appropriate clinical service or the patient’s clinician may be difficult to achieve given the lack of genetics-trained health professionals to meet clinical demand, let alone manage return of research results. Researchers should plan for some flexibility to ensure the cost-effectiveness of RoR [49] and participant education and counselling expenses [48].
Second, researchers should utilise collaborations with clinical experts (e.g., clinical geneticists, genetic counsellors) to develop the RoR plan and return results. Greater clinician support for the return process will increase the perceived utility and clinical usage of results [31].
Third, researchers should be encouraged to share their experiences of the return process, promote best practices through publications, attend conferences to build competence, encourage harmonization of RoR processes across projects and/or biobanks, and share variant data in international databases.
A strength of our approach is that we combined recommendations from existing guidelines with perspectives of European experts across a range of related fields, many of whom have RoR experience. This is important because some existing guidelines were written before much experience had been accumulated. Documents published later than 2020 were excluded from the analysis. However, we note valuable recent contributions to the landscape from Lewis et al. [50] and Willis et al. [51]. In particular, the GA4GH Policy on Clinically Actionable Genomic Research Results supports devising a specific RoR protocol and acquisition of resources for RoR prior to study commencement [50]. Critically, the checklist does not specify a list of genes or specific variants to be returned to participants; this should be determined when study protocols are developed, by a multidisciplinary research team, based on current scientific knowledge and the research context.
Another strength is that utilising European panel members, including legal experts, ensures the checklist is relevant to the European context, as well as more globally. The recommendation to check the three principles articulated in the four legal instruments, as well as country-specific laws [Fig. 1], and a more conservative approach to SF align well with its use in Europe.
While we consider our approach thorough, it is possible some considerations were missed. We encourage researchers to test the checklist by implementing RoR processes into their research projects to identify any gaps. We targeted the checklist to researchers working in research projects/biobanks at their conception, rather than existing studies. Although many of the same considerations pertain when developing RoR processes in existing projects, several aspects, such as obtaining consent, will require consideration.
Data availability
The data from the analysis of the normative documents is available from the corresponding author on request.
Notes
We refer to all potentially returnable findings as IRR unless the guidelines we are referring to specifically state a particular type of IRR.
References
Kerr SM, Klaric L, Halachev M, Hayward C, Boutin TS, Meynert AM, et al. An actionable KCNH2 Long QT Syndrome variant detected by sequence and haplotype analysis in a population research cohort. Sci Rep. 2019;9:1–11.
Leitsalu L, Palover M, Sikka TT, Reigo A, Kals M, Pärn K, et al. Genotype-first approach to the detection of hereditary breast and ovarian cancer risk, and effects of risk disclosure to biobank participants. Eur J Hum Genet. 2021;29:471–81.
Stefansdottir V, Thorolfsdottir E, Hognason HB, Patch C, van El C, Hentze S, et al. Web-based return of BRCA2 research results: one-year genetic counselling experience in Iceland. Eur J Hum Genet. 2020;28:1656–61.
Marjonen H, Marttila M, Paajanen T, Vornanen M, Brunfeldt M, Joensuu A, et al. A Web Portal for Communicating Polygenic Risk Score Results for Health Care Use—The P5 Study. Frontiers in genetics. 2021;12:2170.
Widen E, Junna N, Ruotsalainen S, Surakka I, Mars N, Ripatti P, et al. Communicating polygenic and non-genetic risk for atherosclerotic cardiovascular disease-An observational follow-up study. medRxiv. 2020.
Vears DF, Sénécal K, Clarke AJ, Jackson L, Laberge AM, Lovrecic L, et al. Points to consider for laboratories reporting results from diagnostic genomic sequencing. Eur J Hum Genet. 2018;26:36–43.
Fossey R, Kochan D, Winkler E, Pacyna JE, Olson J, Thibodeau S, et al. Ethical considerations related to return of results from genomic medicine projects: the eMERGE network (phase III) experience. J Personalized Med. 2018;8:2.
Middleton A, Morley KI, Bragin E, Firth HV, Hurles ME, Wright CF, et al. Attitudes of nearly 7000 health professionals, genomic researchers and publics toward the return of incidental results from sequencing research. Eur J Hum Genet. 2016;24:21–9.
Vears DF, Minion JT, Roberts SJ, Cummings J, Machirori M, Blell M, et al. Return of individual research results from genomic research: A systematic review of stakeholder perspectives. PloS one. 2021;16:e0258646.
Mackley MP, Fletcher B, Parker M, Watkins H, Ormondroyd E. Stakeholder views on secondary findings in whole-genome and whole-exome sequencing: a systematic review of quantitative and qualitative studies. Genet Med. 2017;19:283–93.
Dheensa S, Samuel G, Lucassen AM, Farsides B. Towards a national genomics medicine service: the challenges facing clinical-research hybrid practices and the case of the 100 000 genomes project. J Med Ethics. 2018;44:397–403.
Pet DB, Holm IA, Williams JL, Myers MF, Novak LL, Brothers KB, et al. Physicians’ perspectives on receiving unsolicited genomic results. Genet Med. 2019;21:311–8.
Kostick KM, Brannan C, Pereira S, Lázaro‐Muñoz G. Psychiatric genetics researchers’ views on offering return of results to individual participants. Am J Med Genet Part B: Neuropsychiatr Genet. 2019;180:589–600.
Kostick K, Pereira S, Brannan C, Torgerson L, Lázaro-Muñoz G. Psychiatric genomics researchers’ perspectives on best practices for returning results to individual participants. Genet Med. 2020;22:345–52.
Lázaro-Muñoz G, Torgerson L, Smith HS, Pereira S. Perceptions of best practices for return of results in an international survey of psychiatric genetics researchers. Eur J Hum Genet. 2021;29:231–40.
Ferriere M, Van Ness B. Return of individual research results and incidental findings in the clinical trials cooperative group setting. Genet Med. 2012;14:411–6.
Meulenkamp TM, Gevers SJ, Bovenberg JA, Smets E. Researchers’ opinions towards the communication of results of biobank research: a survey study. Eur J Hum Genet. 2012;20:258–62.
Council of Europe. Convention for the protection of individuals with regard to automatic processing of personal data (CETS No. 108). Strasbourg: Council of Europe; 1981. Report No. 9287100225.
Council of Europe. Convention for the protection of individuals with regard to automatic processing of personal data (CETS No. 108). Strasbourg: Council of Europe; 1999. Report No. 9287100225.
Regulation (EU) 2016/679 of the European Parliament and of the Council of 27 April 2016 on the protection of natural persons with regard to the processing of personal data and on the free movement of such data, and repealing Directive 95/46/EC (General Data Protection Regulation), (2016).
Council of Europe. Convention for the protection of Human Rights and Dignity of the Human Being with regard to the Application of Biology and Medicine: Convention on Human Rights and Biomedicine (ETS No. 164). Council of Europe; 1997. Report No. 9287100225.
Council of Europe. Additional Protocol to the Convention on Human Rights and Biomedicine, concerning Biomedical Research (CETS No. 195). Council of Europe; 2005. Report No. 9287100225.
Council of Europe. Explanatory Report to the Additional Protocol to the Convention on Human Rights and Biomedicine, concerning Biomedical Research (CETS No. 195). Council of Europe; 2005. Report No. 9287100225.
Thorogood A, Dalpé G, Knoppers BM. Return of individual genomic research results: are laws and policies keeping step? Eur J Hum Genet. 2019;27:535–46.
Knoppers BM, Joly Y, Simard J, Durocher F. The emergence of an ethical duty to disclose genetic research results: international perspectives. Eur J Hum Genet. 2006;14:1170–8.
Hallowell N, Hall A, Alberg C, Zimmern R. Revealing the results of whole-genome sequencing and whole-exome sequencing in research and clinical investigations: some ethical issues. J Med Ethics. 2015;41:317–21.
Graham M, Hallowell N, Solberg B, Haukkala A, Holliday J, Kerasidou A, et al. Taking it to the bank: the ethical management of individual findings arising in secondary research. J Med Ethics. 2021;47:689.
Henderson GE, Wolf SM, Kuczynski KJ, Joffe S, Sharp RR, Parsons DW, et al. The challenge of informed consent and return of results in translational genomics: empirical analysis and recommendations. J Law Med Ethics. 2014;42:344–55.
Lynch JA, Sharp RR, Aufox SA, Bland ST, Blout C, Bowen DJ, et al. Understanding the return of genomic sequencing results process: Content review of participant summary letters in the eMERGE research network. J Personalized Med. 2020;10:38.
Papaz T, Liston E, Zahavich L, Stavropoulos DJ, Jobling RK, Kim RH, et al. Return of genetic and genomic research findings: experience of a pediatric biorepository. BMC Med Genomics. 2019;12:1–9.
Halverson CM, Bland ST, Leppig KA, Marasa M, Myers M, Rasouly HM, et al. Ethical conflicts in translational genetic research: lessons learned from the eMERGE-III experience. Genet Med. 2020;22:1667–72.
Schwartz ML, McCormick CZ, Lazzeri AL, D’Andra ML, Hallquist ML, Manickam K, et al. A model for genome-first care: returning secondary genomic findings to participants and their healthcare providers in a large research cohort. Am J Hum Genet. 2018;103:328–37.
Aarts N, Bunnik E, Boeckhout M. Guide to the detection, management and communication of incidental findings for biobanks in BBMRI-NL. 2017.
Miller DT, Lee K, Chung WK, Gordon AS, Herman GE, Klein TE, et al. ACMG SF v3. 0 list for reporting of secondary findings in clinical exome and genome sequencing: a policy statement of the American College of Medical Genetics and Genomics (ACMG). Genet Med. 2021;23:1582–4.
Van ElCG, Cornel MC, Borry P, Hastings RJ, Fellmann F, Hodgson SV, et al. Whole-genome sequencing in health care. Eur J Hum Genet. 2013;21:580–4.
de Wert G, Dondorp W, Clarke A, Dequeker E, Cordier C, Deans Z, et al. Opportunistic genomic screening. Recommendations of the European society of human genetics. Eur J Hum Genet. 2021;29:365–77.
European Corporation in Science & Technology. Citizen’s Health through public-private Initiatives: Public health, Market and Ethical perspectives (CHIP ME). 2013.
Budin-Ljøsne I, Mascalzoni D, Soini S, Machado H, Kaye J, Bentzen HB, et al. Feedback of individual genetic results to research participants: is it feasible in Europe? Biopreservation Biobanking. 2016;14:241–8.
Vears DF, Gillam L. Inductive content analysis: A guide for beginning qualitative researchers. Focus Health Professional Educ: A Multi-Professional J 2022;23:111–27.
National Health and Medical Research Council. Chapter 3 - Genomic research. National Statement on Ethical Conduct in Human Research2007 (Updated 2018).
Medical Research Council, Wellcome Trust. Framework on the feedback of health-related findings in research. 2014.
MRCT Center. Return of individual results to participants: Recommendations document. Boston, MA 2017.
National Academies of Sciences E, and Medicine. Returning individual research results to participants: Guidance for a new research paradigm2018.
Landrum MJ, Lee JM, Benson M, Brown GR, Chao C, Chitipiralla S, et al. ClinVar: improving access to variant interpretations and supporting evidence. Nucl Acids Res. 2018;46:D1062–7.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–23.
Presidential Commission for the Study of Bioethical Issues. ANTICIPATE and COMMUNICATE: Ethical Management of Incidental and Secondary Findings in the Clinical, Research, and Direct-to-Consumer Contexts. Washington, DC; 2013.
Samuel GN, Farsides B. The UK’s 100,000 Genomes Project: manifesting policymakers’ expectations. N Genet Soc. 2017;36:336–53.
Zawatsky CLB, Shah N, Machini K, Perez E, Christensen KD, Zouk H, et al. Returning actionable genomic results in a research biobank: Analytic validity, clinical implementation, and resource utilization. Am J Hum Genet. 2021;108:2224–37.
Fontes Marx M, Ataguba JE, Vries JD, Wonkam A. Systematic Review of the Economic Evaluation of Returning Incidental Findings in Genomic Research. Frontiers in. Pub Health. 2021;9:873.
Lewis AC, Knoppers BM, Green RC. An international policy on returning genomic research results. Genome Med. 2021;13:1–3.
Willis AM, Terrill B, Pearce A, McEwen A, Ballinger ML, Young M-A. My Research Results: a program to facilitate return of clinically actionable genomic research findings. Eur J Hum Genet. 2022;30:363–6.
Acknowledgements
We would like to thank Francesca Forzano, Heidi Carmen Howard, and Vigdis Stefánsdóttir for their participation in the workshop and valuable comments on the manuscript. We’d also like to thank our workshop presenters Marcus Perola, Kristian Hveem, Mike Parker, and Neeme Tonisson. Thanks also go to John-Arne Skolbekken for workshop coordination and Fiona Lynch for her assistance with manuscript formatting.
Funding
D.F.V. acknowledges the infrastructure funding received from the Victorian State Government through the Operational Infrastructure Support (OIS) Program. This work was supported by the Australian Government through the Medical Research Future Fund, as part of the Genomics Health Futures Mission (Grant number 76749). N.H. is funded by the Li Ka Shing Foundation. N.H and A.K. are members of the Wellcome Centre for Ethics and Humanities, which is supported by funding from the Wellcome Trust (Grant No. 203132). Work leading up to the guideline development workshop was funded by a Wellcome Trust grant (Returning research results: a Northern European Research Network Grant no 213081/Z/18/Z) awarded to N.H. I.B.L. received funding from Biobank Norway funded by The Research Council of Norway (https://www.forskningsradet.no/en/), grant number 296162/F50. M.T.M. is funded in the context of the activities of BBMRI-ERIC alongside its Work Programme 2021. E.O. is funded by NIHR Oxford Biomedical Research Centre. B.W.S. is supported by Biobank Norway funded through the Research Council of Norway. H.B.B. is funded by NordForsk grant number 81105. S.M.K. is funded by MRC Human Genetics Unit, University of Edinburgh, grant number MC_UU_00007/10, Programme MC_PC_U127592696.
Author information
Authors and Affiliations
Contributions
D.F.V., N.H., and I.B.L. were responsible for the conceptualisation of the study and manuscript. I.B.L. was responsible for the data collection. D.F.V., N.H., and I.B.L. analysed the data. D.F.V. wrote the majority of the first draft of the manuscript with contributions and redrafting from N.H., and I.B.L. H.B.B., B.E., T.H.N., A.K. S.M.K., M.T.M., S.M., E.O., B.S., and B.W.S. participated in the workshop and contributed to the drafting of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
The paper provides a guideline for returning research results developed by the authorship during a policy development workshop. This did not involve any use of personal data or involve human subjects and, therefore, does not require IRB or REC approval.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Vears, D.F., Hallowell, N., Bentzen, H.B. et al. A practical checklist for return of results from genomic research in the European context. Eur J Hum Genet 31, 687–695 (2023). https://doi.org/10.1038/s41431-023-01328-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-023-01328-6
This article is cited by
-
The complex genomics of single gene disorders
European Journal of Human Genetics (2023)
