
Abstract
Jones syndrome is a rare dominantly inherited syndrome characterized by gingival fibromatosis and progressive sensorineural hearing loss becoming symptomatic in the second decade of life. Here, we report a father and his two daughters presenting with a typical Jones syndrome (OMIM %135550) phenotype. Exome sequencing identified a repressor element 1-silencing transcription factor (REST, OMIM *600571) (NM_005612.5) c.2670_2673del p.(Glu891Profs*6) heterozygous variant segregating with Jones syndrome in the family. We review the clinical data from all previously published patients with Jones syndrome and previously published patients with pathogenic REST variants associated with gingival fibromatosis or sensorineural hearing loss. This study suggests that pathogenic REST variants cause Jones syndrome.

Similar content being viewed by others

Introduction
In 1977, Jones et al. described five generations of a family with autosomal dominantly inherited gingival fibromatosis (GF) and progressive sensorineural hearing loss (SNHL) becoming symptomatic late in the second decade of life [1]. The affected individuals had hypertrophied, pink, stippled, and nodular gingiva, predominantly in the anterior part of the mouth and predominant premaxillae. Clinical and audiometric examinations revealed progressive downward-sloping SNHL. The syndrome was named Jones syndrome (OMIM %135550), and several families with Jones syndrome were later described in the medical literature [2,3,4,5]. However, the underlying gene causing Jones syndrome remains elusive.
Pathogenic variants in the repressor element 1-silencing transcription factor (REST) gene (OMIM *600571) have previously been associated with dominantly inherited SNHL [6, 7], GF [8,9,10], or susceptibility to Wilms tumor [11]. Here, we describe a Finnish family with a typical Jones syndrome phenotype. Exome sequencing identified a pathogenic REST variant segregating with the phenotype. To our knowledge, this is the first report of the association between a pathogenic REST variant and Jones syndrome.
Subjects and methods
Molecular genetics
Whole-exome sequencing (WES) was performed for Patients 1–3 in Centogene (Rostock, Germany). Targeted Sanger sequencing was used for segregation analysis of the identified REST variant. Details of sequencing and bioinformatics are included in the supplement.
Clinical data
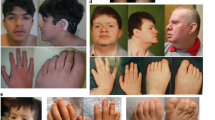
Patient 1 is an 11-year-old female (Supplementary Fig. 1, II-5). After birth, transient evoked otoacoustic emissions were not obtained, but an automated auditory brainstem response was obtained bilaterally. Her growth and development were normal. At age 10, she was referred for further investigations due to GF prolonging the retention of primary teeth and delaying permanent tooth eruption (Fig. 1A). Dental panoramic tomography (DPT) showed that all the unerupted teeth were present and had normal root development (Fig. 1C), and lateral cephalometric radiograph showed a convex profile with an open bite tendency (Fig. 1D). Extractions of over-retained primary teeth and the removal of excessive gingiva covering non-erupted permanent teeth were performed to ensure the eruption of the teeth (Fig. 1B). An audiogram revealed mild bilateral SNHL affecting the high frequencies (Fig. 1E). She does not need hearing aids and has mild hypertrichosis and hypertelorism.

A. Patient 1 at age 10 with marked gingival fibromatosis, which prolonged the retention of primary teeth and delayed permanent tooth eruption. B. Gingiva of Patient 1 after surgical treatment. C. A pantomogram of Patient 1 showing that all the unerupted teeth were present with normal root development. D. A cephalogram of Patient 1 showing a convex profile with an open bite tendency. Audiograms of Patient 1 at age 11 (E.) and Patient 2 at age 14 (F) show mild bilateral SNHL affecting the high frequencies. Audiograms of Patient 3 at age 5 (G) and age 42 (H) show the progression of SNHL from initially affecting only the high frequencies to gradually affecting the middle frequencies as well.
Patient 2, an older sister of Patient 1, is a 14-year-old female. Her growth and development were normal. At the age of five years, mild bilateral SNHL affecting the high frequencies was identified (Fig. 1F). No remarkable progression has been noted, and she does not need hearing aids. She has GF, which has delayed permanent tooth eruption. The DPTs showed normal root development, with delayed eruption because of primary retention. Cephalometric analysis showed sagittal jaw discrepancy with neutral facial growth. She also has mild hypertrichosis.
Patient 3, the father of Patients 1–2, is a 42-year-old male, whose parents and siblings do not have SNHL or GF. At the age of one year, bilateral inguinal hernias were corrected surgically. At age four, right-sided Legg-Perthes-Calve disease was treated with osteotomy. His bone age was delayed. He has mild hypertrichosis. At age five, he was diagnosed with bilateral SNHL affecting predominantly high frequencies (Fig. 1G), and his hearing loss has slowly progressed. His latest audiograms at age 40 revealed SNHL affecting the middle and high frequencies (Fig. 1H). He is using hearing aids. At age ten, he was referred to a dentist due to markedly delayed (2.5 years) permanent tooth eruption. At age 14, he was referred to an orthodontist due to a convex profile with a retrognathic mandible, and labially inclined upper incisors with an excessive overjet and open bite. There was also generalized GF preventing the eruption of several permanent teeth. He was treated with orthodontics as a teenager, and a gingivectomy was performed at age 20. At follow-up, GF had not recurred.
Results
Whole-exome sequencing
WES of Patients 1–3 identified a heterozygous REST (NM_005612.5) c.2670_2673del p.(Glu891Profs*6) (GRCh38 NC_000004.12:g.56931528_56931531del) variant (Supplementary Fig. 2). Sanger sequencing of the unaffected siblings confirmed the segregation (Supplementary Fig. 1). The variant is not present in the Genome Aggregation Database (v.2.1.1). It is classified as pathogenic (PVS1, PM2, PP1) according to the ACMG criteria [12].
Review of the literature
The clinical and genetic details of the REST-related phenotypes are summarized in Table 1 and pathogenic REST variants reported in the literature are shown in Supplementary Fig. 3.
Previously published patients with Jones syndrome
Twelve patients with Jones syndrome have been described previously (Supplementary Table 1) [1,2,3,4,5]. All had GF, which was often severe, and five required surgical treatment. Histology of the extracted gingiva showed thickened epithelium, elongated rete ridges, inflammation, and dense fibrous connective tissue stroma. Ten had SNHL, and audiogram results often moderate to severe SNHL affecting mainly higher frequencies. The average age of onset for SNHL was 14 years (10–20 years), and for GF 9 years (4–20 years). Other findings included hirsutism, hypermobility of the distal phalanges, hypertrophic scar, undescended testis, and intellectual disability.
Pathogenic REST variants causing gingival fibromatosis
An original report described nine individuals from three families with GF caused by three different pathogenic REST final-exon-truncating variants [8]. Chen et al. reported two patients with GF caused by two novel truncating pathogenic REST variants [9]. In vitro, reported gene assays demonstrated that these truncated RESTs had a partial or complete loss of repressor activity and that truncated RESTs impaired the repressive ability of the wild-type REST, suggesting a dominant negative effect [9]. Recently, Machado et al. reported a novel truncating REST variant segregating with mild GF in a Brazilian family [10].
Pathogenic REST variants causing sensorineural hearing loss
Peters et al. mapped a dominantly inherited postlingual progressive SNHL to the REST-containing DFNA27 locus in chromosome 4 [13]. Sequencing of all annotated exons in the DFNA27 locus failed to identify potentially pathogenic variants, but sequencing of conserved intronic regions of REST revealed a heterozygous intronic variant segregating with a hearing loss phenotype in the family [7]. This intronic REST variant (NM_005612.5) c.983-2247C>T resulted in the prevention of alternative splicing of the REST mRNA, which was essential for the regulation of REST in the inner ear [7]. Manyisa et al. reported on a South African family presenting with progressive non-syndromic SNHL caused by the REST c.1244G>C, p.(Cys415Ser) missense variant [6]. The affected individuals in these two families showed either a flat-type audiogram affecting all frequencies or a downward-sloping audiogram affecting mostly the high frequencies.
Discussion
This brief report describes a father and his two daughters presenting with GF and progressive SNHL, also known as Jones syndrome. WES identified a small deletion in the last exon of REST, segregating with the phenotype.
REST encodes a transcriptional regulator expressed in most non-neuronal cells and its expression is minimal in maturing hair cells and most neurons [7]. It contains a DNA- binding domain that can bind to a specific 21 base pair consensus sequence (RE-1) in the promoter region of its target genes to repress the transcription of the target genes [14]. REST acts as a master negative regulator of neurogenesis, a regulator of osteoblast differentiation, and a key repressor of gene expression in hypoxia [15].
Previously, pathogenic REST variants were associated with progressive dominantly inherited SNHL in two families [6, 7]. The affected individuals in these two families had either a flat-type audiogram affecting all the frequencies or downward-sloping audiograms, similar to Patients 1–3 described in this report. REST is also involved in the organization of the auditory receptor cell stereocilium [15]. REST expression was decreased in hair cells and spiral ganglion neurons in age-related hearing loss in mice. REST has a protective role in age-related hearing loss, and its deficiency upregulates p53 and induces cochlear cell apoptosis, which then leads to SNHL [14].
The TGF-β/Smad signaling cascade and overproduction of the extracellular matrix (ECM), especially collagen type I, have been proposed to account for GF [16, 17]. REST is expressed in the epithelium and lamina propria of normal and fibrotic gingiva [9], and it controls gingival homeostasis by supressing profibrotic genes and activating proteolytic genes [9]. It is speculated that loss-of-function REST variants cause increased production but reduced turnover of the ECM leading to GF [10].
Previously, a deep intronic REST c.983-2247C>T variant has been demonstrated to cause dysregulation of alternative splicing and deafness and a gain-of-function has been suggested as a disease mechanism [7]. This variant is not present in any of the three affected individuals from this study, and therefore it can be excluded as causing SNHL in our patients. Recent studies in mice seem to contradict the gain-of-function hypothesis as deletion of REST in the cochlea resulted in hearing impairment and induced apoptosis of spiral ganglion neurons and hair cells [14], suggesting that loss of REST function may increase the susceptibility to SNHL. REST c.2670_2673del is in the last exon and leads to protein truncation probably escaping nonsense-mediated decay suggesting that susceptibility to SNHL and gingival hypertrophy may be caused by a loss-of-function mechanism. Also, a dominant negative mechanism has been suggested as a disease mechanism in patients with pathogenic REST variants [9]. However, we cannot exclude the possibility that SNHL in our family would be caused by a yet unidentified pathogenic variant.
It is possible that patients reported in ref. [6,7,8,9,10] actually had Jones syndrome as SNHL and gingival hypertrophy may be mild or even non-penetrant [1] and not routinely sought after in patients with gingival or audiological phenotypes. While further studies are required to confirm the association between REST and Jones syndrome, based on our study, we recommend detailed oral and hearing examinations of patients with potentially pathogenic heterozygous REST variants. As SNHL in young patients can be relatively mild, predominantly affecting high frequencies and progressing slowly, it may be undiagnosed without proper audiological testing.
Jones syndrome is a rare, possibly underdiagnosed, slowly progressive disease. Early diagnosis and follow-up allow timely treatment of GF and hearing loss, decreasing the severity of the disease and improving patients’ quality of life.
Data availability
The reported variant was submitted to the LOVD database hosted at Leiden University Medical Center, the Netherlands (DB-ID REST_000030).
References
Jones G, Wilroy RS, McHaney V. Familial gingival fibromatosis associated with progressive deafness in five generations of a family. Birth Defects Orig Artic Ser. 1977;13:195–201.
Da R, Singh S, Gupta I, Gopal S. Gingival enlargement in a case of variant Jones Syndrome: a case report. J Dent. 2016;17:62–6.
Gita B, Chandrasekaran S, Manoharan P, Dembla G. Idiopathic gingival fibromatosis associated with progressive hearing loss: a nonfamilial variant of Jones syndrome. Contemp Clin Dent. 2014; https://doi.org/10.4103/0976-237X.132387.
Hartsfield JK, Bixler D, Hazen RH. Gingival fibromatosis with sensorineural hearing loss: an autosomal dominant trait. Am J Med Genet. 1985; https://doi.org/10.1002/ajmg.1320220323.
Kasaboğlu O, Tümer C, Balci S. Hereditary gingival fibromatosis and sensorineural hearing loss in a 42-year-old man with Jones syndrome. Genet Couns. 2004;15:213–8.
Manyisa N, Schrauwen I, de Souza Rios LA, Mowla S, Tekendo-Ngongang C, Popel K, et al. A monoallelic variant in REST is associated with non-syndromic autosomal dominant hearing impairment in a South African family. Genes. 2021; https://doi.org/10.3390/genes12111765.
Nakano Y, Kelly MC, Rehman AU, Boger ET, Morell RJ, Kelley MW, et al. Defects in the alternative splicing-dependent regulation of REST cause deafness. Cell 2018;174:536–548. https://doi.org/10.1016/j.cell.2018.06.004.
Bayram Y, White JJ, Elcioglu N, Cho MT, Zadeh N, Gedikbasi A, et al. RESt final-exon-truncating mutations cause hereditary gingival fibromatosis. Am J Hum Genet. 2017;101:149–156. https://doi.org/10.1016/j.ajhg.2017.06.006.
Chen JT, Lin CH, Huang HW, Wang YP, Kao PC, Yang TP, et al. Novel REST truncation mutations causing hereditary gingival fibromatosis. J Dent Res. 2021;100:868–874. https://doi.org/10.1177/0022034521996620.
Machado RA, de Andrade RS, Pêgo SPB, Krepischi ACV, Coletta RD, Martelli-Júnior H. New evidences of genetic heterogeneity of causing hereditary gingival fibromatosis and ALK and CD36 as new candidate genes. J Periodontol. 2022; https://doi.org/10.1002/JPER.22-0219.
Mahamdallie SS, Hanks S, Karlin KL, Zachariou A, Perdeaux ER, Ruark E, et al. Mutations in the transcriptional repressor REST predispose to Wilms tumor. Nat Genet. 2015;47:1471–4. https://doi.org/10.1038/ng.3440.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24. https://doi.org/10.1038/gim.2015.30.
Peters LM, Fridell RA, Boger ET, San Agustin TB, Madeo AC, Griffith AJ, et al. A locus for autosomal dominant progressive non-syndromic hearing loss, DFNA27, is on chromosome 4q12-13.1. Clin Genet. 2008;73:367–72. https://doi.org/10.1111/j.1399-0004.2008.00966.x.
Li H. Downregulation of REST in the cochlea contributes to age-related hearing loss via the p53 apoptosis pathway. Cell Death Dis. 2022;13:343 https://doi.org/10.1038/s41419-022-04774-0.
UniProt Consortium. UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Res. 2021;49:D480–D489. https://doi.org/10.1093/nar/gkaa1100.
Roman-Malo L, Bullon B, de Miguel M, Bullon P. Fibroblasts collagen production and histological alterations in hereditary gingival fibromatosis. Diseases. 2019;7:39 https://doi.org/10.3390/diseases7020039.
Sobral LM, Montan PF, Zecchin KG, Martelli-Junior H, Vargas PA, Graner E, et al. Smad7 blocks transforming growth factor-β1-induced gingival fibroblast-myofibroblast transition via inhibitory regulation of Smad2 and connective tissue growth factor. J Periodontol. 2011;82:642–51. https://doi.org/10.1902/jop.2010.100510.
Funding
This study was funded by Academy of Finland (grant number 338446), Uolevi Mäki Foundation, and the Competitive State Research Financing of the Expert Responsibility Area of Oulu University Hospital (grant number VTR K36733). We thank the family who participated in this study. Open Access funding provided by University of Oulu including Oulu University Hospital.
Author information
Authors and Affiliations
Contributions
Conceptualization: MKT and ER; Writing—original draft: MKT, JJ and ER; Writing—review and editing: MKT, JJ, SK, TK, SW, AMBA, SH, and ER.
Corresponding author
Ethics declarations
Competing interests
SW, AMBA are employees of CENTOGENE GmbH. The authors declare no conflicts of interest.
Ethics
Written informed consent was obtained from all the patients and unaffected siblings participating in the study. Written informed consent was obtained to publish patient photos. The study was approved by the Ethics Committee of the Northern Ostrobothnia Hospital District (Eettmk § 186, 17/08/2020).
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rahikkala, E., Julku, J., Koskinen, S. et al. Pathogenic REST variant causing Jones syndrome and a review of the literature. Eur J Hum Genet 31, 469–473 (2023). https://doi.org/10.1038/s41431-022-01258-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-022-01258-9
This article is cited by
-
2023 in the European Journal of Human Genetics
European Journal of Human Genetics (2024)
-
April, again
European Journal of Human Genetics (2023)
-
The cause of Jones syndrome put to REST: a mutation in the REST gene causes gingival fibromatosis and hearing loss
European Journal of Human Genetics (2023)
