
Abstract
The yield of chromosomal microarray analysis (CMA) is well established in structurally normal fetuses (0.4–1.4%). We aimed to determine the incremental yield of exome sequencing (ES) in this population. From February 2017 to April 2022, 1,526 fetuses were subjected to ES; 482 of them were structurally normal (31.6%). Only pathogenic and likely pathogenic (P/LP) variants, per the American College of Medical Genetics and Genomics (ACMG) classification, were reported. Additionally, ACMG secondary findings relevant to childhood were reported. Four fetuses (4/482; 0.8%) had P/LP variants indicating a moderate to severe disease in ATP7B, NR2E3, SPRED1 and FGFR3, causing Wilson disease, Enhanced S-cone syndrome, Legius and Muenke syndromes, respectively. Two fetuses had secondary findings, in RET and DSP. Our data suggest that offering only CMA for structurally normal fetuses may provide false reassurance. Prenatal ES mandates restrictive analysis and careful management combined with pre and post-test genetic counseling.
Similar content being viewed by others

Introduction
Exome sequencing (ES) is a powerful tool for identifying disease-causing single nucleotide variants (SNVs) and small insertions and deletions. Its diagnostic yield among individuals suspected of being affected by a genetic condition is reported at around 31–48% [1, 2] and even higher in highly inbred populations [3]. In the prenatal setup, the incremental diagnostic yield of ES in large non-selected cohorts of fetuses suspected of having a genetic condition is estimated at around 8.5–10%, depending on the referral indication, but is reported to be as high as 30% in a few meta-analyses [4,5,6,7].
As compared to ES, which was designed for single base-pair resolution, chromosomal microarray analysis (CMA) were designed to detect copy number variants (deletions and duplications) at a resolution of dozens to hundreds of thousands of base pairs. The yield of CMA in fetuses without a specific indication is around 0.4–1.4% [8,9,10] (as compared to a yield of 4–10% in fetal malformations [11,12,13]), reflecting the fact that genetic disorders might not be sonographically detected at the various stages of pregnancy. Thus, the policy of performing prenatal testing using CMA upon “parental request” has become widely accepted [8,9,10, 14]. Nontheless, it is clear that normal CMA analysis does not preclude an underlying genetic condition stemming from a SNV, below CMA detection limit.
ES utilities markedly outgrow those offered by CMA; ES can detect severe diseases caused by SNVs, by small CNVs that are below CMA resolution [15] and uniparental heterodisomy in addition to isodisomy in trios [16]. Moreover, it can also replace CMA in the detection of large CNVs [17]. Therefore, we analyzed the yield of ES in structurally normal fetuses, by conducting a retrospective study in this population of fetuses who underwent ES per parental request (Fig. 1A).

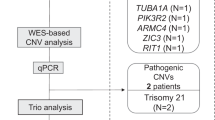
A The yield of CMA and exome sequencing in prenatal diagnosis. The yield of CMA refers to the ratio of positive test after normal karyotype. The yield of ES refers to the ratio of positive tests after a negative CMA. The relevant references are cited in the manuscript. CMA chromosomal microarray analysis, ES Exome sequencing. B Schematic presentation of the study design and findings. *A positive result (pathogenic and likely pathogenic variants, classified according to severity of disease) was classified according to Lazarin et al. [19] Secondary findings were analyzed according to the ACMG guidelines [18]. According to the ACMG guidelines, were reported in with a moderate to severe disease expected. ACMG American College of Medical Genetics and Genomics. SF Secondary findings, ES Exome sequencing, P pathogenic, LP likely pathogenic.
Methods
Study design
This is a retrospective study of all prenatal ES analyses of structurally normal fetuses in a tertiary center in Jerusalem, Israel, between the years 2017 and 2022. The term “sonographically normal” is related to the date of prenatal sampling. A pretest genetic counseling session took place before the test performance. Only pathogenic and likely pathogenic (P/LP) variants, classified according to American College of Medical Genetics and genomics (ACMG) guidelines, in genes causing moderate to severe diseases were reported. In addition, ACMG secondary findings that are relevant to health during childhood or found in a proband-only ES were reported [18]. Parents provided written informed consent for ES analysis following a thorough genetic counseling session.
Inclusion and exclusion criteria
The study included only fetuses without any medical indication, who underwent CMA and were referred to ES due to parental wish and had no pathogenic or likely pathogenic (P/LP) CNV. Fetuses with abnormal sonographic/first/second trimester testing results, P/LP CMA findings (either as separate CMA result or by CNV analysis of the ES data), or a known parental illness were excluded.
Exome sequencing
Following informed consent, ES was performed on DNA extracted from amniocytes or chorionic villi and from parental peripheral blood. It was performed either as a proband-only, duo, trio or quatro design (fetus/es and parent/s). Capture and sequencing methods as well as details of downstream analyses are provided as Supplementary Methods.
Results
From February 2017 to April 2022, 9861 fetuses underwent prenatal diagnosis. Some 60% of them are known to be structurally normal [14]. 1526 fetuses underwent ES in this time period and 482 of them were structurally normal. There were 235 single ES (48.8%), 214 sets of trio (44.4%), 15 sets of duo (3.1%), and18 quads (3.7%). 48 couples (~10%) opted for ES due to a previously affected child with a pathogenic de novo variant. Thirteen families (2.7%) requested the analysis due to advanced paternal age and increased risk for de novo mutation, and eight couples (1.6%) opted for ES due to consanguinity and an increased risk for autosomal recessive disorders. The remaining group (of 413 families) was enriched for medical care professionals (as per personal communication).
ES results were classified into two categories, “Positive” and “Negative”, based on the report of a P/LP variant. Positive results were further sub-divided into “moderate” and “severe”, based on the severity of disease, and according to Lazarin et al. [19] (Fig. 1B). Disclosure policy precluded diseases of mild severity. ACMG incidental secondary findings were reported in case of childhood onset.
The overall positive result was 1.24%. In four of 482 cases (0.83%), a positive result for a moderate to severe disease was reported: two fetuses carried compound heterozygous variants in ATP7B and NR2E3, associated with Wilson disease and with retinal disorder, respectively. Another fetus carried a de novo pathogenic variant in SPRED1, known to cause Legius syndrome and another harbored a de novo pathogenic variant in FGFR3 related to Muenke syndrome. As for ACMG incidental secondary findings, two positive findings were found (0.4%). One fetus had a paternally-inherited pathogenic variant in RET (not known to the father until then), a known susceptibility gene for childhood onset malignancies (Table 1), and one fetus was found to carry a heterozygous pathogenic variant in DSP, a known cause of arrhythmogenic right ventricular cardiomyopathy. Segregation studies were recommended, to determine if the DSP variant was inherited and to provide surveillance recommendations for the relevant parent.
Two fetuses with a 16p13.11 microdeletion and a Klinefelter Syndrome (47, XXY), were excluded from the statistics. No UPD events were identified. None of the positive fetuses were among the consanguineous families, yet only eight couples were consanguineous. Results were conveyed to the couples in a comprehensive genetic counseling session. Parents of fetuses harboring pathogenic variants in the genes ATP7B, NR2E3, SPRED1 and FGFR3 opted for pregnancy termination. Pregnancies of the fetuses with ACMG secondary findings, harboring pathogenic variants in RET and DSP, continued to term.
Discussion
Our retrospective study identified positive ES results in 0.83 of structurally normal fetuses. Two fetuses inherited biallelic pathogenic variants in ATP7B and NR2E3, causing recessively-inherited Wilson disease (OMIM #277900) and enhanced S-cone disease (OMIM #268100), respectively. All four variants were reported in the literature as pathogenic. Wilson disease is a disorder of copper metabolism that can present with hepatic, neurologic, psychiatric disturbances, or a combination of these, in individuals ranging from age 3 years to older than 50 years. Early treatment with copper chelating agents or zinc in asymptomatic individuals may prevent manifestations [20]. Receiving such a diagnosis for a fetus exemplifies the rationale behind performing fetal ES, i.e., earning invaluable information about the fetus that can help parents prevent a severe disease or initiating early pre-symptomatic treatment. Enhanced S-cone or retinitis pigmentosa are retinal diseases that may be caused by biallelic pathogenic variants in NR2E3 [21]. Although gene therapy for retinal diseases is emerging [22], there is currently no postnatal prevention or cure for this specific subtype. Notably, an exome-based carrier-screening program for recessive disorders would have identified parental shared carrier status in both instances. This highlights the importance of an expanded carrier-screening test [23] in couples who wish to minimize chances for an affected child, either by gene panel or exome-duo. The variant in SPRED1 gene causes Legius syndrome (AKA Neurofibromatosis Type 1-Like Syndrome, OMIM # 611431). The characteristics of this disease are the presence of multiple café au lait macules, intertriginous freckling, lipomas, macrocephaly and variable neurodevelopmental delay. The disease-causing variant was not inherited. Muenke syndrome is caused by the variant c.749C>G (p.Pro250Arg) in FGFR3. Its variable expression may include coronal synostosis of various types, hearing loss, developmental delay, epilepsy, intracranial anomalies and intellectual disability [24, 25]. The penetrance is incomplete but high (OMIM # 602849). Enrichment of P/LP de novo variants in individuals with neurodevelopmental disorders is well established [26,27,28]. Advanced paternal age at conception has been associated with an increased risk of autosomal dominant genetic disorders [29,30,31]. Some of these paternal age effect mutations are positively selected and lead to relative enrichment of mutant sperm over time [32]. The frequency of autosomal dominant disease due to de novo variants when the paternal age is 40 years or older at conception is estimated to be as high as 0.5% [33, 34]. These data, combined with the two pathogenic de novo variants detected in this study, suggest that offering ES due to advanced paternal age could be beneficial.
Notably, 10% of the couples in our cohort opted for ES due to a previously affected child with a pathogenic de novo variant. Whereas these couples were eligible for testing for the specific identified variant in every subsequent pregnancy due to the risk of germline mosaicism, they opted for a comprehensive test. Since these parents had received genetic counseling focusing on this mode of inheritance, they may have been particularly aware that other de novo variants can occur and that CMA testing would not be able to detect them. Furthermore, some of these couples are parenting severely ill children and were therefore invested in minimizing the risk of giving birth to another affected child.
One fetus had a paternally-inherited pathogenic variant in RET (not known to the father until then), a known susceptibility gene for childhood onset malignancies (Table 1), and one fetus was found to carry a paternally-inherited heterozygous pathogenic variant in DSP, a known cause of arrhythmogenic right ventricular cardiomyopathy [35]. The latter had a significant family history of fainting and cardiac death. The RET variant reported in this study was a paternally-inherited pathogenic variant known to be associated with multiple endocrine neoplasia type 2 (OMIM #171400, 162300), yet without any known clinical consequences in the family. Since RET-related malignancies may appear early in life [36], and it is medically actionable, disclosure is recommended by the ACMG guidelines [18]. In such cases, a clear benefit could be obtained for the fetus and the transmitting parent. DSP encodes a desmosomal protein important for tissue integrity. Pathogenic variants in DSP cause arrhythmogenic right ventricular dysplasia/cardiomyopathy (ARVD) [37]. This is an inherited cardiac disease, clinically characterized by electrical abnormalities and high frequency of re-entry arrhythmias, mainly originating from the right ventricle, which may lead to palpitations, syncope, and even sudden death at a young age. Results would be life-saving in case the variant is inherited, since cardiac follow-up should be recommended to carriers.
Our disclosure policy that included only moderate and severe diseases might be challenged and must be emphasized during the pretest genetic counseling session. Diseases of mild severity (i.e., familial Mediterranean fever or factor XI deficiency) may have incomplete penetrance and variable expression without a clear genotype-phenotype correlation [38]. The benefit of reporting these diseases in terms of early surveillance and treatment should be weighed against the risk of initiating undue stress during pregnancy. In recent years we recognized the necessity of genetic counseling for every woman/couple opting for prenatal diagnosis, especially to discuss the robustness of genomic tests in the prenatal setup and uncertainties which may arise. We believe that in the genomic era, this should be the policy in every center performing prenatal diagnosis. Exposing parents to a mild to moderate fetal disease in the prenatal rather than the postnatal period is certainly stressful and might direct them to pregnancy termination or challenge their bonding with the newborn. Issues of diseases with variable expression or reduced penetrance should be thoroughly discussed with parents opting for prenatal ES. Not surprisingly, some parents may decide to forgo this modality in order to avoid situations of making decisions regarding pregnancy outcome while facing an equivocal diagnosis.
There is only one report in the literature regarding ES yield in structurally normal fetuses amounting to 1/160 (0.6%) [39]. The relatively high yield in our cohort might be attributed to several biases, including a small sample size and a high prevalence of founder mutations in the local populations. One of the main drawbacks of ES is equivocal variant classification [40]. Nonetheless, variant classification methodologies have greatly improved in the past years due to both internal variant databases that enable calculation of population-specific minor allele frequency (MAF), as well as publicly available databases such as GnomAD (https://gnomad.broadinstitute.org/), and ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/). Hence the accuracy of variant classification is constantly increasing. On the other hand, the limited classification due to lack of an observed phenotype in seemingly healthy fetuses poses a challenge and may lead to an underestimation of positive cases. ACMG Standards and Guidelines recommend 28 criteria for classifying variant pathogenicity. Three criteria are based on the clinical phenotype of the index case (PP4, PS2, PP1) [40] and thus are automatically excluded from the classification of structurally normal fetuses. The limited phenotypic delineation of the fetus turns molecular diagnoses with variable expression and incomplete penetrance to burdensome and stressful, since decisions regarding pregnancy’s outcome are based upon limited information. These aspects of ES should be discussed in the pretest counseling thoroughly.
The subject of termination along different stages of pregnancy is a complicated moral, ethical, religious and legal issue. While in most Europe states terminations beyond the first trimester are more complicated legally, in Canada there is no timing limitation during pregnancy. In Israel, every termination of pregnancy should be approved by a special committee which use more strict criteria after the 24th week, when the fetus is considered viable. The performance of ES for a structurally normal fetus at early second trimester, may reveal a severe disease early enough to terminate pregnancy before viability.
In Israel, ES during pregnancy is paid out of pocket without funding by the ministry of health or the different health maintenance organizations. This is in sharp contrast to CMA which is funded by both authorities. In view of the present report, we propose that this policy should be changed. ES includes all diagnostic yield of CMA and provide additional findings in a considerable fraction of the seemingly healthy fetuses. In the near future ES or whole genome sequencing might gradually become the first-tier test in prenatal diagnosis, with data regarding SNV, CNV, aneuploidy and more obtained by this single platform. This data can also be used for the benefit of the fetus/newborn and his/her parents when actionable secondary findings are highlighted. Thus, a treatable genetic disease diagnosed in utero can save precious time in initiating therapy. Cautious interpretation and an established disclosure policy are critical, along with extensive pre and post-test genetic counseling. Although this is the largest cohort, larger studies are called for in order to establish the incremental yield of ES beyond CMA and to further discuss the medical and ethical challenges of prenatal ES in structurally normal fetuses.
Data availability
Data will be available following a reasonable request. Variants were submitted to ClinVar (SUB9513879). Accession numbers are SCV001572879.1, SCV001572880.1, SCV001572881.1, SCV001572882.1, SCV001572883.1, SCV001437667.1, SCV001437666.1.
Code availability
The variants described in the article can be found in Table 1. Additional data are available from the corresponding author on reasonable request.
References
Al-Dewik N, Mohd H, Al-Mureikhi M, Ali R, Al-Mesaifri F, Mahmoud L, et al. Clinical exome sequencing in 509 Middle Eastern families with suspected Mendelian diseases: the Qatari experience. Am J Med Genet A. 2019;179:927–35.
Lee H, Deignan JL, Dorrani N, Strom SP, Kantarci S, Quintero-Rivera F, et al. Clinical exome sequencing for genetic identification of rare Mendelian disorders. JAMA. 2014;312:1880–7.
Hengel H, Buchert R, Sturm M, Haack TB, Schelling Y, Mahajnah M, et al. First-line exome sequencing in Palestinian and Israeli Arabs with neurological disorders is efficient and facilitates disease gene discovery. Eur J Hum Genet. 2020;28:1034–43.
Lord J, McMullan DJ, Eberhardt RY, Rinck G, Hamilton SJ, Quinlan-Jones E, et al. Prenatal exome sequencing analysis in fetal structural anomalies detected by ultrasonography (PAGE): a cohort study. Lancet. 2019;393:747–57.
Petrovski S, Aggarwal V, Giordano JL, Stosic M, Wou K, Bier L, et al. Whole-exome sequencing in the evaluation of fetal structural anomalies: a prospective cohort study. Lancet. 2019;393:758–67.
Mellis R, Oprych K, Scotchman E, Hill M, Chitty LS. Diagnostic yield of exome sequencing for prenatal diagnosis of fetal structural anomalies: a systematic review and meta-analysis. Prenat Diagn. 2022;42:662–85.
Pauta M, Martinez-Portilla RJ, Borrell A. Diagnostic yield of exome sequencing in fetuses with multisystem malformations: systematic review and meta-analysis. Ultrasound Obstet Gynecol. 2022;59:715–22.
Stern S, Hacohen N, Meiner V, Yagel S, Zenvirt S, Shkedi-Rafid S, et al. Universal chromosomal microarray analysis reveals high proportion of copy number variants in low risk pregnancies. Ultrasound Obstet Gynecol. 2021;57:813–20.
Sagi-Dain L, Cohen Vig L, Kahana S, Yacobson S, Tenne T, Agmon-Fishman I, et al. Chromosomal microarray vs. NIPS: analysis of 5541 low-risk pregnancies. Genet Med. 2019;21:2462–7.
Moshonov R, Hod K, Azaria B, Abadi-Korek I, Berger R, Shohat M. Benefit versus risk of chromosomal microarray analysis performed in pregnancies with normal and positive prenatal screening results: a retrospective study. PLoS ONE. 2021;16:e0250734.
Wapner RJ, Martin CL, Levy B, Ballif BC, Eng CM, Zachary JM, et al. Chromosomal microarray versus karyotyping for prenatal diagnosis. N Engl J Med. 2012;367:2175–84.
Zhang Z, Hu T, Wang J, Hu R, Li Q, Xiao L, et al. Pregnancy outcomes of fetuses with congenital heart disease after a prenatal diagnosis with chromosome microarray. Prenat Diagn. 2022;42:79–86.
Tzadikevitch Geffen K, Singer A, Maya I, Sagi-Dain L, Khayat M, Ben-Shachar S, et al. Chromosomal microarray should be performed for cases of fetal short long bones detected prenatally. Arch Gynecol Obstet. 2021;303:85–92.
Hochner H, Daum H, Douiev L, Zvi N, Frumkin A, Macarov M, et al. Information women choose to receive about prenatal chromosomal microarray analysis. Obstet Gynecol. 2020;135:149–57.
Pfundt R, Del Rosario M, Vissers LELM, Kwint MP, Janssen IM, de Leeuw N, et al. Detection of clinically relevant copy-number variants by exome sequencing in a large cohort of genetic disorders. Genet Med. 2017;19:667–75.
Yauy K, de Leeuw N, Yntema HG, Pfundt R, Gilissen C. Accurate detection of clinically relevant uniparental disomy from exome sequencing data. Genet Med. 2020;22:803–8.
Kadalayil L, Rafiq S, Rose-Zerilli MJ, Pengelly RJ, Parker H, Oscier D, et al. Exome sequence read depth methods for identifying copy number changes. Brief Bioinform. 2015;16:380–92.
Kalia SS, Adelman K, Bale SJ, Chung WK, Eng C, Evans JP, et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): a policy statement of the American College of Medical Genetics and Genomics. Genet Med. 2017;19:249–55.
Lazarin GA, Hawthorne F, Collins NS, Platt EA, Evans EA, Haque IS. Systematic classification of disease severity for evaluation of expanded carrier screening panels. PLoS ONE. 2014;9:e114391.
Dzieżyc K, Karliński M, Litwin T, Członkowska A. Compliant treatment with anti-copper agents prevents clinically overt Wilson’s disease in pre-symptomatic patients. Eur J Neurol. 2014;21:332–7.
Hull S, Arno G, Sergouniotis PI, Tiffin P, Borman AD, Chandra A, et al. Clinical and molecular characterization of enhanced S-cone syndrome in children. JAMA Ophthalmol. 2014;132:1341–9.
Ferraz Sallum JM, Godoy J, Kondo A, Kutner JM, Vasconcelos H, Maia A. The first gene therapy for RPE65 biallelic dystrophy with voretigene neparvovec-rzyl in Brazil. Ophthalmic Genet. 2022;1–5. Online ahead of print.
Kraft SA, Duenas D, Wilfond BS, Goddard KAB. The evolving landscape of expanded carrier screening: challenges and opportunities. Genet Med. 2019;21:790–7.
Öwall L, Kreiborg S, Dunø M, Hermann NV, Darvann TA, Hove H. Phenotypic variability in Muenke syndrome-observations from five Danish families. Clin Dysmorphol. 2020;29:1–9.
González-Del Angel A, Estandía-Ortega B, Alcántara-Ortigoza MA, Martínez-Cruz V, Gutiérrez-Tinajero DJ, Rasmussen A, et al. Expansion of the variable expression of Muenke syndrome: hydrocephalus without craniosynostosis. Am J Med Genet A. 2016;170:3189–96.
Järvelä I, Määttä T, Acharya A, Leppälä J, Jhangiani SN, Arvio M, et al. Exome sequencing reveals predominantly de novo variants in disorders with intellectual disability (ID) in the founder population of Finland. Hum Genet. 2021;140:1011–29.
Monies D, Abouelhoda M, Assoum M, Moghrabi N, Rafiullah R, Almontashiri N, et al. Lessons learned from large-scale, first-tier clinical exome sequencing in a highly Consanguineous population. Am J Hum Genet. 2019;104:1182–201.
Veltman JA, Brunner HG. De novo mutations in human genetic disease. Nat Rev Genet. 2012;13:565–75.
Zhytnik L, Peters M, Tilk K, Simm K, Tõnisson N, Reimand T, et al. From late fatherhood to prenatal screening of monogenic disorders: evidence and ethical concerns. Hum Reprod Update. 2021;27:1056–85.
Yatsenko AN, Turek PJ. Reproductive genetics and the aging male. J Assist Reprod Genet. 2018;35:933–41.
Orioli IM, Castilla EE, Scarano G, Mastroiacovo P. Effect of paternal age in achondroplasia, thanatophoric dysplasia, and osteogenesis imperfecta. Am J Med Genet. 1995;59:209–17.
Goriely A, McGrath JJ, Hultman CM, Wilkie AO, Malaspina D. “Selfish spermatogonial selection”: a novel mechanism for the association between advanced paternal age and neurodevelopmental disorders. Am J Psychiatry. 2013;170:599–608.
Friedman JM. Genetic disease in the offspring of older fathers. Obstet Gynecol. 1981;57:745–9.
Janecka M, Mill J, Basson MA, Goriely A, Spiers H, Reichenberg A, et al. Advanced paternal age effects in neurodevelopmental disorders-review of potential underlying mechanisms. Transl Psychiatry. 2017;7:e1019.
Groeneweg JA, van der Zwaag PA, Olde Nordkamp LR, Bikker H, Jongbloed JD, Jongbloed R, et al. Arrhythmogenic right ventricular dysplasia/cardiomyopathy according to revised 2010 task force criteria with inclusion of non-desmosomal phospholamban mutation carriers. Am J Cardiol. 2013;112:1197–206.
Makri A, Akshintala S, Derse-Anthony C, Widemann B, Stratakis CA, Glod J, et al. Multiple endocrine neoplasia type 2B presents early in childhood but often is undiagnosed for years. J Pediatr. 2018;203:447–9.
Lazzarini E, Jongbloed JD, Pilichou K, Thiene G, Basso C, Bikker H, et al. The ARVD/C genetic variants database: 2014 update. Hum Mutat. 2015;36:403–10.
Ben-Chetrit E. Familial Mediterranean fever (FMF) and renal AA amyloidosis-phenotype-genotype correlation, treatment and prognosis. J Nephrol. 2003;16:431–4.
Vaknin N, Azoulay N, Tsur E, Tripolszki K, Urzi A, Rolfs A, et al. High rate of abnormal findings in Prenatal Exome Trio in low risk pregnancies and apparently normal fetuses. Prenat Diagn. 2022;42:725–35.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24.
Acknowledgements
We would like to thank the genetic counselors and laboratory team for a professional teamwork, and the families for their participation in the study.
Funding
There was no financial support in this work.
Author information
Authors and Affiliations
Contributions
Conceptualization—HMS, HD, TH, Formal analysis—HMS, OE, VM, TM, Resources—CR, AE, DF, Software—SGN, AB, Writing—original draft—HMS, HD, Writing—review and editing—TH, NY, SP, DK, SY, TM, DVV.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
This project was approved by the IRB and listed 0189-21-HMO. Any information reported is de-identified. An informed consent was obtained from all participants as required by the IRB.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Daum, H., Harel, T., Millo, T. et al. Exome sequencing for structurally normal fetuses—yields and ethical issues. Eur J Hum Genet 31, 164–168 (2023). https://doi.org/10.1038/s41431-022-01169-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-022-01169-9
This article is cited by
-
2023 in the European Journal of Human Genetics
European Journal of Human Genetics (2024)
-
The value of exomes across the ages
European Journal of Human Genetics (2023)
