
Abstract
Neurofibromatosis type 1 (NF1), an autosomal dominant disorder characterized by skin pigmentary lesions and multiple cutaneous neurofibromas, is caused by neurofibromin 1 (NF1) loss of function variants. Currently, a molecular diagnosis is frequently established using a multistep protocol based on cDNA and gDNA sequence analysis and/or Multiplex Ligation-dependent Probe Amplification (MLPA) assay on genomic DNA, providing an overall detection rate of about 95–97%. The small proportion of clinically diagnosed patients, which at present do not obtain a molecular confirmation likely are mosaic, as their pathogenic variant may remain undetected due to low sensitivity of low coverage NGS approaches, or they may carry a type of pathogenic variant refractory to currently used technologies. Here, we report two unrelated patients presenting with two different inversions that disrupt the NF1 coding sequence, resulting in an NF1 phenotype. In one subject, the inversion was associated with microdeletions spanning a few NF1 exons at both breakpoints, while in the other the rearrangement did not cause exon loss, thus testing negative by MLPA assay. Considering the high proportion of repeated regions within the NF1 sequence, we propose that intragenic structural rearrangements should be considered as possible pathogenic mechanisms in patients fulfilling the NIH diagnostic criteria of NF1 but lacking of molecular confirmation and in patients with NF1 intragenic microdeletions.
Similar content being viewed by others
Introduction
Neurofibromatosis type 1 (NF1, OMIM # 16220) is an autosomal dominant disorder resulting from loss of function variants in NF1 gene. Its worldwide incidence is about 1 in 2000 while its prevalence varies greatly by country, also depending on diagnostic possibilities, population age distribution and access to health care [1]. The disease is gradually progressive, clinically characterized by skin pigmentary lesions (café-au-lait spots, skinfold freckling and Lisch nodules) and multiple cutaneous neurofibromas. Less frequent but potentially more serious features include brain and peripheral nerve tumors (optic nerve and other central nervous system gliomas, malignant peripheral nerve sheath tumors), skeletal abnormalities (tibial dysplasia, scoliosis), learning disability and behavioral problems. NF1 is fully penetrant with a highly variable expression, even within the same family.
An international consensus has been recently published [2], defining minimal clinical and genetic criteria for NF1 and Legius syndrome, which can present with overlapping features in young patients.
Nevertheless, clinical diagnosis can be challenging in non-familial cases due to heterogeneity of clinical manifestations and their age-related occurrence. Indeed, when applying the diagnostic criteria to an infant, it should be kept into consideration that nodular and plexiform neurofibromas can develop later on and may not be present at the time of the clinical diagnosis. Only a few genotype-phenotype correlations have been established so far [3,4,5,6,7,8].
NF1 gene codifies for neurofibromin, a tumor suppressor protein, expressed ubiquitously, acting as a key negative regulator of the RAS-MAPK signaling pathway. Therefore, NF1 loss of function is predicted to upregulate cell growth and survival, increasing the active GTP-bound form of RAS. Several pathogenic mechanisms have been reported. While germinal heterozygous loss of function is considered causative for some constitutional features, biallelic inactivation through a somatic second hit is required for café-au-lait spots, neurofibromas development, tibial dysplasia and malignant peripheral nerve sheath tumors [9].
NF1 has one of the highest known mutation rates among human genetic diseases, with about half of the cases being sporadic. Genomic sequence variants account for 60–90% of the cases [10] and nearly 3000 different nucleotide variants are presently annotated as pathogenic in The Human Gene Mutation Database (HGMD, Institute of Medical Genetics, Cardiff, http://www.hgmd.cf.ac.uk/ac/index.php), including nonsense, missense, splicing, microdeletions, insertions, indel and complex rearrangements [11]. About 12.5% of patients harbor a microdeletion or a microduplication detectable by Multiplex Ligation-dependent Probe Amplification (MLPA) specific assay or Chromosomal Microarray Analysis [4]. These can include one or more NF1 exons, the whole gene sequence or larger regions involving NF1 and the neighboring genes (OMIM# 613675) [12]. Structural chromosomal rearrangements interrupting the NF1 sequence have been described and are estimated to account for about 1% of the cases [13,14,15,16,17]. In particular, a recurrent nonrandom 17;22 translocation was reported [11, 18, 19], mediated by long palindromic AT-rich regions (PATRRs) with GC-rich ends within intron 31 of NF1.
Molecular diagnosis of NF1 is based on Multistep pathogenic variant detection protocol based on cDNA and gDNA analysis, providing an overall detection rate of about 95–97% [20, 21]. Patients with no SNV detected after NGS analysis are usually analyzed for deletion/duplication by an MLPA test. Despite the high detection rate of these laboratory procedures, about 3–5% of clinically diagnosed cases do not find a molecular confirmation [20], probably due to tissue mosaicisms or different mutational events, such as structural rearrangements [11, 19] unsolved by standard techniques.
Optical genome mapping is a new non-sequencing genome imaging tool able to detect copy number and structural variants at high resolution [22, 23]. It relies on the purification of ultra-high molecular weight (UHMW) DNA on which labels are attached via a non-destructive direct label and stain (DLS) technology. The resulting ultra-long DNA molecules, bearing this uniquely identifiable genome-specific label patterns, are directly imaged and used for building an accurate physical genome map. Comparative analysis of the label patterns over long contiguous reads across the whole genome, reveals the occurrence of both copy number variants and structural variants.
Here we report two patients with a clinical diagnosis of NF1, harboring microinversions disrupting the coding sequence of NF1. Our results delineate structural rearrangements as a possible pathogenic mechanism responsible for the NF1 phenotype.
Material and methods
Clinical data were obtained in accordance with the ethical standards of the Bambino Gesù Children Hospital (Rome, Italy). Informed consent was signed by the patients’ parents. The study protocol conforms to the ethical guidelines of the 1975 Declaration of Helsinki, as reflected in a priori approval by the institution’s Human Research Committee. The patients were clinically evaluated at the Medical Genetics Unit of the Hospital.
Patient 1 had clinical diagnosis of NF1, based on the association of sparse multiple cafe-au-lait spots, axillary and inguinal freckling, cutaneous neurofibromas on the back, the right shoulder, lower limbs and left foot, and bilateral Lisch nodules. Cognitive development was normal. At age of 11.8 years, weight was 40.500 kg (75th centile), height 148.5 cm (50–75th centile), head circumference 55 cm (75th centile).
Patient 2 presented clinical features diagnostic for NF1, including multiple cutaneous café-au-lait spots, axillary and inguinal freckling, multiple cutaneous neurofibromas on the latero-cervical left region, three neurofibromas on the right retroauricular region, a single neurofibroma on the left palm (removed and histologically examined), joint laxity, dorso-lumbar scoliosis. Additional features included arching of the anterior leaflet of the mitral valve resulting in mild valve insufficiency, umbilical hernia (operated), hypertelorism, thick lips, overfolded helices. Cognitive development was normal. At age 17.8 years weight was 54.400 kg (10th centile), height 171.4 cm (25–50th centile), head circumference 55 cm (25th centile).
Molecular analysis
Genomic DNA of probands and their parents was isolated from peripheral blood by a QIAsymphony automatic extractor (QIAGEN, www.qiagen.com) using standard procedures.
Next generation sequencing
Library preparation was carried out according to the manufacturer’s protocol (Twist Bioscience), and sequenced on a NovaSeq6000 (Illumina) platform. The target parameters were the coding exons including a region extension of 25 bases from the 3′ end and 25 bases from the 5′ end (based on RefSeq database). We obtained a targeted NGS assay that had a mean 150× coverage for >97% bases, a specificity and sensitivity of 100%, with a quality score of ≥30. The BaseSpace pipeline and the TGex software LifeMap Sciences were used for variant calling and annotation. Sequencing data were aligned to the hg19 human reference genome. Based on the guidelines of the American College of Medical Genetics and Genomics, a minimum depth coverage of 30× was considered suitable for analysis. Mutational analysis was performed by a custom “virtual” panel comprehensive of all genes associated to NF1 and related disorders.
Multiplex ligation-dependent probe analysis
MLPA was performed by using the SALSA MLPA kit P081 and P082 containing probes for each exon of NF1 gene, available from MRC Holland (MRC-Holland, Amsterdam, The Netherlands). Analysis was performed by electrophoretic run on a 3130xl automatic sequencer (Applied Biosystems by Life Technologies). Results were analyzed by a dedicated software (Coffalyser.Net).
Optical Genome Mapping (OGM) and structural variant calling
Optical genome mapping is a non-sequencing genome imaging tool for high-speed, high-throughput copy number and structural variant detection and analysis with high sensitivity and specificity.
A fresh blood sample was collected in EDTA and stored at −80 °C just after sampling. UHMW DNA was extracted according to the manufacturer’s instructions (SP Frozen Human Blood DNA Isolation Protocol, Bionano Genomics), and enzymatically labeled by the DLE-1 Enzyme (Bionano Prep Direct Label and Stain Protocol). It is a non-destructive DLS technology, leaving DNA samples intact.
A volume of 8.5 µl of labeled gDNA solution of concentration between 4 and 12 ng/µl was loaded on Saphyr chip and scanned on the Saphyr instrument (Bionano genomics, San Diego USA). Saphyr chip were ran to reach a minimum yield of 320 Gbp corresponding to 100× effective coverage. Saphyr chips are composed of hundreds of thousands of parallel NanoChannels, enabling high-throughput data acquisition to build an accurate Bionano genome map. Each de novo assembled map corresponds to the consensus pairwise alignment of the labeled DNA molecules imaged on the Saphyr instrument. Comparative analysis of the label patterns over long contiguous reads across the whole genome, reveals copy number variants (>500 Kb) and structural variants (>30 Kp), at high sensitivities and specificity [24].
The de novo assembly and Variant Annotation Pipeline were executed on Bionano Solve software V3.6 using Human Genome Reference Consortium GRCh38 assembly as a reference for structural variants detection. Reporting and direct visualization of structural variants was done on Bionano Access V1.6.
Whole-genome sequencing (WGS)
WGS was performed on genomic DNA in order to provide a fine characterization of the inversions breakpoints. Library preparation was carried out according to the manufacturer’s protocol from DNA PCR-Free Library Prep (Illumina), and sequenced on a NovaSeq6000 (Illumina) platform. The obtained NGS assay presented a mean coverage of 35×, with Q30 bases around 87%. The TruSight Software Suite (illumina) and the integrated DRAGEN platform and IGV software were used for alignment, variant calling and breakpoint data visualization. Sequencing data were aligned to the hg38 human reference genome.
Results
NGS analysis was performed on both patients according to the standard diagnostic protocol for NF1. No causative variants were detected in NF1 or related genes.
In Patient 1, NF1 locus specific MLPA analysis showed two distinct noncontiguous microdeletions, involving exons 4–7 and 31–35, respectively. Both microdeletions arose de novo, suggesting the presence of an underlying structural variant. In Patient 2, MLPA assay tested negative.
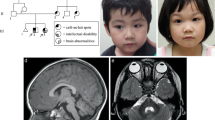
OGM analysis, performed in both patients, revealed the presence of distinct inversions disrupting the coding sequence of NF1. In Patient 1, an intragenic 68.5 Kb inversion, spanning from NF1 exon 8 to exon 30, was detected, flanked by the two microdeletions located at the proximal and distal breakpoints (Fig. 1). In Patient 2, a larger balanced inversion was found, with the distal breakpoint located within intron 3 of NF1 gene, and the proximal one 499 Kb upstream in a gene desert region (Fig. 2). Both the intervals where the two breakpoints are located are rich in repeated sequences: SINE (Short Interspersed Nuclear Elements), LINE (Long Interspersed Nuclear Elements), LTR (Long Terminal Repeats).

a MLPA analysis detects two distinct noncontiguous microdeletions involving exons 4–7 and 31–35 respectively of NF1 gene. b OGM analysis discloses the presence of a 68.5 Kb intragenic inversion having the two microdeletions as breakpoints. The patient optical map is aligned and compared with the reference optical map. Molecular labels are reported as vertical lines on both patient’s and reference’s maps.

The patient optical map is aligned and compared with the reference optical map. Molecular labels are reported as vertical lines on both patient’s and reference’s maps. VA was responsible for data analysis and writing the report. FRL was responsible for data analysis and writing the report. MLD was responsible for patients’ clinical evaluation and manuscript revision. SG was responsible for data analysis, literature and paper revision. ES was responsible for data analysis. KB technical support for genome wide analysis. BD was responsible for revising and approving the paper. RC was responsible for patients’ clinical evaluation and paper revision. AN was responsible for revising and approving the paper. MCD was responsible for patients’ clinical evaluation and paper revision.
In order to determine the genomic coordinates of the breakpoints, a whole-genome sequencing analysis was performed (WGS), allowing a fine characterization of the two variants.
Patient 1: NC_000017.11:g.[31162374_ 31182520del;31182521_31250996inv;31250997_ 31266506del]
patient 2: NC_000017.11:g.30660608_31159168inv
A further investigation of the involved sequences has not been performed and the underlying mechanisms predisposing to the rearrangement was not determined.
Discussion
NF1 is a large 287 Kb in size gene, with 57 constitutive and three alternatively spliced exons [25]. Mutation detection is challenging due to the large size of the gene, the wide allelic heterogeneity, the lack of mutational hotspots, the presence of unprocessed pseudogenes, and the occurrence of mosaic tissue specific variants [21, 26]. About 3–5% of the cases do not receive a molecular confirmation by using standard multistep protocols, usually based on NGS, MLPA/microarray and cytogenetic analysis.
OGM detects both copy number variants (deletion and duplication at an average resolution >500 bp) and structural variants (>30 Kb), providing detailed information about the rearrangements’ breakpoints. It also provides a specific and unique labeling pattern on double strand long DNA molecules, allowing the detection of balanced and unbalanced rearrangements at an extremely high resolution.
We report on two unrelated patients presenting with a clinical diagnosis of NF1, harboring structural microrearrangements affecting the coding sequence of NF1 detected by OGM and missed by the standard of care.
In Patient 1, two de novo microdeletions, involving exons 4–7 and 31–35, have been detected using a specific MLPA assay. Although the microdeletions were classified as pathogenic, thus confirmatory of the clinical diagnosis, they were regarded as unlikely independent constitutional events, suggesting the presence of an underlying structural variant, which was confirmed by OGM analysis disclosing an intragenic inversion with breakpoints at the microdeletion sites. Two additional cases have been described, presenting with a double intragenic deletion. Wimmer et al. [27] reported a NF1 patient with two noncontiguous microdeletions, involving exons 31 and 33–35, while Pasmant et al. [25] described an individual with two microdeletions in exons 32–36 and 49–58. Considering the high mutational rate of NF1, the occurrence of two independent mutational events cannot be excluded a priori. However, the presence of an underlying structural event, in particular an inversion, explaining the co-occurrence of deletions deserves consideration as a pathogenic mechanism for NF1.
In Patient 2, both NGS and MLPA tested negative and a molecular diagnosis was reached by means of OGM. In fact, a balanced microinversion, 499 Kb in size, was shown to interrupt the coding sequence of NF1 at intron 1. To the best of our knowledge, no similar patient heterozygous for a balanced microinversion of NF1 has been reported so far.
Both the inversions found in our patients were too small to be detected by standard karyotype or FISH analysis, and therefore were missed by standard diagnostic protocols. Unfortunately, RNA from peripheral blood of these patients was not available for testing and the possible transcription of the rearranged gene was not evaluated. However, while a rearranged RNA could be expected for patient 1, we consider unlikely that the inversion detected in patient 2 could lead to a transcription product. In fact, only one of the breakpoints fall within NF1 sequence (intron 1) while the other one is located 499 kb upstream, reallocating NF1 promotor and the first 3 exons several kilobases apart from the remaining portion of the gene. Therefore, it is likely to assume that cDNA sequencing in patient 2 would test negative as well.
The OGM technology may provide a molecular diagnosis in a fraction of patients in whom routine diagnostic protocols did not identify the causative pathogenic variant and may contribute to an estimate of the frequency of structural microarrangements in NF1.
A molecular confirmation of NF1 is important for addressing an appropriate surveillance especially for an early detection of malignant tumors, collecting accurate data for related clinical trials, and developing targeted therapies [9]. In addition, mainly in children and in patients with atypical presentation, it is important to differentiate NF1 from diseases manifesting overlapping features, such as Legius syndrome, skin hyperpigmentation, mismatch repair and overgrowth syndromes. Finally, a confirmatory molecular diagnosis is crucial for genetic counseling, assessment of the reproductive risk and possible prenatal diagnosis.
In conclusion, we provide evidence and focus the attention on structural microrearrangements as possible damaging events leading to Neurofibromatosis type 1, in association or not with microdeletions. Additional studies are needed to assess the frequency of these structural changes, which could be overlooked by standard NF1 diagnostic protocols.
Data availability
Data generated during this study have been submitted in ClinVar (ID SUB11349933, SUB11349931).
References
Kallionpää RA, Uusitalo E, Leppävirta J, Pöyhönen M, Peltonen S, Peltonen J. Prevalence of neurofibromatosis type 1 in the Finnish population. Genet Med. 2018;20:1082–6.
Legius E, Messiaen L, Wolkenstein P, Pancza P, Avery RA, Berman Y, et al. Revised diagnostic criteria for neurofibromatosis type 1 and Legius syndrome: an international consensus recommendation. Genet Med. 2021;23:1506–13.
Upadhyaya M, Huson SM, Davies M, Thomas N, Chuzhanova N, Giovannini S, et al. An absence of cutaneous neurofibromas associated with a 3-bp inframe deletion in exon 17 of the NF1 gene (c.2970-2972 delAAT): evidence of a clinically significant NF1 genotype-phenotype correlation. Am J Hum Genet. 2007;80:140–51.
Smith MJ, Urquhart JE, Harkness EF, Miles EK, Bowers NL, Byers HJ, et al. The contribution of whole gene deletions and large rearrangements to the mutation spectrum in inherited tumor predisposing syndromes. Hum Mutat. 2016;37:250–6.
Rojnueangnit K, Xie J, Gomes A, Sharp A, Callens T, Chen Y, et al. High incidence of Noonan syndrome features including short stature and pulmonic stenosis in patients carrying NF1 missense mutations affecting p.Arg1809: genotype-phenotype correlation. Hum Mutat. 2015;36:1052–63.
Ruggieri M, Polizzi A, Spalice A, Salpietro V, Caltabiano R, D’Orazi V, et al. The natural history of spinal neurofibromatosis: a critical review of clinical and genetic features. Clin Genet. 2015;87:401–10.
Koczkowska M, Callens T, Chen Y, Gomes A, Hicks AD, Sharp A, et al. Clinical spectrum of individuals with pathogenic NF1 missense variants affecting p.Met1149, p.Arg1276, and p.Lys1423: genotype-phenotype study in neurofibromatosis type 1. Hum Mutat. 2020;41:299–315.
Koczkowska M, Chen Y, Callens T, Gomes A, Sharp A, Johnson S, et al. Genotype-phenotype correlation in NF1: evidence for a more severe phenotype associated with missense mutations affecting NF1 codons 844-848. Am J Hum Genet. 2018;102:69–87.
Gutmann DH, Ferner RE, Listernick RH, Korf BR, Wolters PL, Johnson KJ. Neurofibromatosis type 1. Nat Rev Dis Prim. 2017;3:17004.
Calì F, Chiavetta V, Ruggeri G, Piccione M, Selicorni A, Palazzo D, et al. Mutation spectrum of NF1 gene in Italian patients with neurofibromatosis type 1 using Ion Torrent PGM™ platform. Eur J Med Genet. 2017;60:93–9.
Smith RB, Solem EP, Metz EC, Wheeler FC, Phillips JA 3rd, Yenamandra A. Clinical diagnosis of neurofibromatosis type I in multiple family members due to cosegregation of a unique balanced translocation with disruption of the NF1 locus: Testing considerations for accurate diagnosis. Am J Med Genet A. 2021;185:1222–7.
Kehrer-Sawatzki H, Mautner VF, Cooper DN. Emerging genotype-phenotype relationships in patients with large NF1 deletions. Hum Genet. 2017;136:349–76.
Ledbetter DH, Rich DC, O’connell P, Leppert M, Carey JC. Precise localization of NF1 to 17q11. 2 by balanced translocation. Am J Hum Genet. 1989;44:20–4.
Menon AG, Ledbetter DH, Rich DC, Seizinger BR, Rouleau GA, Michels VF, et al. Characterization of a translocation within the von Recklinghausen neurofibromatosis region of chromosome 17. Genomics. 1989;5:245–9.
Asamoah A, North K, Doran S, Wagstaff J, Ogle R, Collins FS, et al. 17q inversion involving the neurofibromatosis type one locus in a family with neurofibromatosis type one. Am J Med Genet. 1995;60:312–6.
Messiaen LM, Callens T, Mortier G, Beysen D, Vandenbroucke I, Van Roy N, et al. Exhaustive mutation analysis of the NF1 gene allows identification of 95% of mutations and reveals a high frequency of unusual splicing defects. Hum Mutat. 2000;15:541–55.
Schmidt MA, Michels VV, Dewald GW, Opitz JM, Reynolds JF. Cases of neurofibromatosis with rearrangements of chromosome 17 involving band 17q11.2. Am J Med Genet. 1987;28:771–7.
Kehrer-Sawatzki H, Häussler J, Krone W, Bode H, Jenne DE, Mehnert KU, et al. The second case of a t(17;22) in a family with neurofibromatosis type 1: sequence analysis of the breakpoint regions. Hum Genet. 1997;99:237–47.
Kurahashi H, Shaikh T, Takata M, Toda T, Emanuel BS. The constitutional t(17;22): another translocation mediated by palindromic AT-rich repeats. Am J Hum Genet. 2003;72:733–8.
Evans DG, Bowers N, Burkitt-Wright E, Miles E, Garg S, Scott-Kitching V, et al. Comprehensive RNA analysis of the NF1 gene in classically affected NF1 affected individuals meeting NIH criteria has high sensitivity and mutation negative testing is reassuring in isolated cases with pigmentary features only. EBioMedicine. 2016;7:212–20.
Valero MC, Martín Y, Hernández-Imaz E, Marina Hernández A, Meleán G, Valero AM, et al. A highly sensitive genetic protocol to detect NF1 mutations. J Mol Diagn. 2011;13:113–22.
Mantere T, Neveling K, Pebrel-Richard C, Benoist M, van der Zande G, Kater-Baats E, et al. Optical genome mapping enables constitutional chromosomal aberration detection. Am J Hum Genet. 2021;108:1409–22.
Sabatella M, Mantere T, Waanders E, Neveling K, Mensenkamp AR, van Dijk F, et al. Optical genome mapping identifies a germline retrotransposon insertion in SMARCB1 in two siblings with atypical teratoid rhabdoid tumors. J Pathol. 2021;255:202–11.
Hastie AR, Lam ET, Pang AWC, Zhang LX, Andrews W, Lee J, et al. Rapid automated large structural variation detection in a diploid genome by nanochannel based next-generation mapping. BioRxiv. 2017. https://doi.org/10.1101/102764.
Pasmant E, Parfait B, Luscan A, Goussard P, Briand-Suleau A, Laurendeau I, et al. Neurofibromatosis type 1 molecular diagnosis: what can NGS do for you when you have a large gene with loss of function mutations? Eur J Hum Genet. 2015;23:596–601.
Messiaen LM, Wimmer K. NF1 mutational spectrum. In: Kaufmann D, editor. Neurofibromatoses, Vol. 16. Basel: Karger; 2008, pp, 63–77.
Wimmer K, Yao S, Claes K, Kehrer-Sawatzki H, Tinschert S, De Raedt T, et al. Spectrum of single- and multiexon NF1 copy number changes in a cohort of 1,100 unselected NF1 patients. Genes Chromosomes Cancer. 2006;45:265–76.
Funding
No financial assistance was received in support of the study.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval
The study protocol conforms to the ethical guidelines of the 1975 Declaration of Helsinki, as reflected in a priori approval by the institution’s Human Research Committee.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Alesi, V., Lepri, F.R., Dentici, M.L. et al. Intragenic inversions in NF1 gene as pathogenic mechanism in neurofibromatosis type 1. Eur J Hum Genet 30, 1239–1243 (2022). https://doi.org/10.1038/s41431-022-01153-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-022-01153-3
This article is cited by
-
Analysis of complex chromosomal rearrangement involving chromosome 6 via the integration of optical genomic mapping and molecular cytogenetic methodologies
Journal of Human Genetics (2024)
-
OGM and WES identifies translocation breakpoints in PKD1 gene in an polycystic kidney patient and healthy baby delivered using PGT
BMC Medical Genomics (2023)
-
Comment on Intragenic inversions in NF1 gene as pathogenic mechanism in neurofibromatosis type 1
European Journal of Human Genetics (2023)
-
Genome sequencing—do you know what you are getting into?
European Journal of Human Genetics (2022)
