
Abstract
Intellectual disability (ID), a neurodevelopmental disorder affecting 1–3% of the general population, is characterized by limitations in both intellectual function and adaptive skills. The high number of conditions associated with ID underlines its heterogeneous origin and reveals the difficulty of obtaining a rapid and accurate genetic diagnosis. However, the Next Generation Sequencing, and the whole exome sequencing (WES) in particular, has boosted the diagnosis rate associated with ID. In this study, WES performed on 244 trios of patients clinically diagnosed with isolated or syndromic ID and their respective unaffected parents has allowed the identification of the underlying genetic basis of ID in 64 patients, yielding a diagnosis rate of 25.2%. Our results suggest that trio-based WES facilitates ID’s genetic diagnosis, particularly in patients who have been extensively waiting for a definitive molecular diagnosis. Moreover, genotypic information from parents provided by trio-based WES enabled the detection of a high percentage (61.5%) of de novo variants inside our cohort. Establishing a quick genetic diagnosis of ID would allow early intervention and better clinical management, thus improving the quality of life of these patients and their families.
Similar content being viewed by others


Introduction
Intellectual disability (ID), a neurodevelopmental disorder with a prevalence of 1–3% in the general population, is characterized by significant intellectual and adaptive functioning limitations with onset in the developmental period [1]. ID can show a variable degree of severity –ranging from mild to profound– and can be classified as non-syndromic (presented in isolation) or syndromic (associated with other clinical features) [2]. Moreover, ID is highly comorbid with epilepsy, motor abnormalities, psychiatric symptoms, or autism spectrum disorder (ASD) [3, 4]. Among ID’s highly heterogeneous underlying genetic causes, it is worth mentioning chromosomal alterations, copy number variants (CNVs) or deleterious variants in single genes [5]. In this sense, the monogenic forms of ID could follow an autosomal dominant (AD), autosomal recessive (AR) or X-linked (XL) pattern of inheritance [6,7,8]. Interestingly, it is estimated that around 2000 genes would be involved in the different inheritance patterns of ID [9,10,11].
ID supposes a great personal effort for both affected individuals and their families and entails important implications regarding the support, necessary care, and special education that these patients require. Early recognition of ID patients would immediately implement actions to improve their cognitive and adaptive skills [12, 13]. Over the last few decades, many genetic tests have been developed to identify the genetic alterations associated with ID, including karyotype, fluorescent in situ hybridization, single-gene Sanger sequencing, and chromosomal microarray analysis [14, 15]. However, ineffective genetic testing usually prolongs the diagnostic odyssey for years [16, 17]. It is common to find adults among the reported cohorts from several recent studies, despite that in most of them the symptomatology was evident from an early age [18, 19]. The advent of Next Generation Sequencing (NGS) technologies, and particularly the Whole Exome Sequencing (WES), has led to a widespread increase in the gene discovery and diagnosis rate associated with ID [12, 20,21,22,23,24]. Thus, whereas the estimated diagnostic yield for ID in the well-established targeted NGS gene panel was ~21% (up to 39% using trio-based panels), this rate reached values between ~25% and ~55% in WES [16, 19, 21, 25,26,27]. Nevertheless, WES’s diagnostic rate would be further increased through family trio sequencing [17, 28,29,30,31]. Its proven ability to identify new candidate genes or variants in known genes, low processing time, increasingly high accuracy, and excellent cost-effectiveness convert WES into an optimum ID diagnosis approach [32, 33]. This strategy could facilitate the rapid and efficient identification of causal variants in sporadic cases, in patients with unaffected parents and no family history, or in patients where both parents show a similar phenotype [17].
In this study, through trio-WES analysis performed in a cohort of 254 ID patients, we were able to identify pathogenic (P) or likely pathogenic (LP) variants in 64 of them, which yields an overall diagnostic rate of 25.2%. Moreover, a high rate of de novo variants (DNVs) was noted inside this cohort. These findings seem to extend the proven trio-WES’s utility for diagnosing ID. In this regard, an early genetic diagnosis of ID could provide an adequate intervention and clinical management of these patients.
Material and methods
Patient recruitment
In total, 244 trios of non-consanguineous healthy parents and affected probands were recruited from several Galician associations, most of them under the auspices of FADEMGA (Federación gallega de asociaciones en favor de las personas con discapacidad intelectual o del desarrollo); from the Pediatric Neurology Departments at Galician hospitals; and from the Fundación Pública Galega de Medicina Xenómica. In seven of these trios, there were two or more affected siblings. All 254 selected patients have a clinical diagnosis of ID made by trained pediatric neurologists or psychiatrists based on the Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition Text Revision and Fifth Edition (DSM-IV-TR and DSM-5) criteria. 54 patients (21.26%) have a mild grade of ID, 39 (15.35%) a moderate grade, 29 (11.42%) a severe grade, and in 132 (51.97%) the grade has not been specified. Normal results were obtained from array CGH and FMR1 CGG-repeat status in 65 (26.4%) and 11 (4.3%) out of the 254 analyzed patients, respectively (Supplementary Table S2).
Whole exome sequencing
DNA was isolated from peripheral blood of the 254 patients using the GentraPuregene blood kit (Qiagen Inc., Valencia, CA, USA), following manufacturer’s instructions. In total, 50 µL of each genomic DNA sample (at a concentration of 80 ng/µL) were sent to the Autism Sequencing Consortium (ASC; https://genome.emory.edu/ASC/) to perform trio-WES on Illumina HiSeq sequencers using the Illumina Nextera exome capture kit [34]. A large multisample VCF containing the raw data obtained from WES was retrieved from the ASC.
Data processing and annotation
Samples were selected from a large multisample VCF file using BCFtools, extracting only variants with PASS information in the FILTER column, splitting and annotating them individually to preserve individual variant information from the variant caller. Annotation was performed by ANNOVAR using several hg38 databases, thus obtaining for each variant its genetic RefSeq based annotation, population frequency (1000 Genomes Project, gnomAD, dbSNP), clinical information (OMIM, HGMD, ClinVar, GeneReviews, CGD, and InterVar), functional prediction (everything recorded in dbNSFP such as SIFT, Polyphen, CADD, PhyloP, GERP + + , and others) or splicing prediction (dbscSNV and regSNP). Individual annotations were then filtered by RefSeq’s coding positions using BEDTools, discarding those variants located more than 10 bp from both gene ends. After this, we constructed a merged VCF file containing the variants from each proband and respective parents. Finally, we filtered the merged files with a virtual gene panel. However, we have also analyzed the full exome data to detect variants in genes not so far associated with ID.
Sample tracing control
A sample tracing protocol was performed by evaluating the alleles present at 24 genomic positions internally selected. Samples were genotyped using Sequenom MassARRAY® multiplex genotyping platform (Sequenom, Inc., San Diego, CA) for such 24 genomic positions. Results were compared with those obtained by sequencing, uniquely assigning sequencing results to their origin samples. All samples included in our study passed this tracing control.
Virtual panel
To make the genetic test more approachable and faster, we have designed a virtual panel of genes combining the available information from the Genomics England PanelApp database (https://panelapp.genomicsengland.co.uk/; accessed on 02/09/2020) regarding panels associated with ID, ASD, and epilepsy. After selecting the genes with green or amber color code and discarding those with red code, the number of genes included in our virtual panel was 1810 (Supplementary Table S1). However, to avoid any loss of information during the VCF filtering according to the genes contained in the virtual panel, we decided to include all known aliases of these initially selected genes in the virtual panel. Therefore, our virtual panel reaches a final number of 6879 items.
Variant filtering, interpretation, and classification
We expect to reduce the large number of variants identified by WES through variant filtering, prioritizing those variants that could be of potential interest in each patient. For this, cut-offs were adequately settled to avoid including false positives and excluding true positive variants. Several filters were applied to both the complete VCF files and the virtual panel’s output files obtained from filtering. This variant filtering pipeline was based on the following steps:
-
1.
Filtering by allelic frequency. We selected variants whose maximum value of minimum allele frequency (MAF) in any population group of the data shared by gnomAD (https://gnomad.broadinstitute.org/) or The 1000 Genomes Project (https://www.internationalgenome.org/1000-genomes-browsers/) was ≤0.05%. All others were discarded.
-
2.
Filtering by predicted consequence on protein level. We selected missense, nonsense, frameshift (insertion and deletion), non-frameshift (insertion and deletion), synonymous (with potential harmful effect over splicing), stop-loss, start-loss, and splice site disruption variants. Missense variants reported as Benign or Likely benign in ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) or InterVar (http://wintervar.wglab.org/) databases, or with CADD score <15 or DANN score <0.93 were discarded. DNA variants inside 5ʹ or 3ʹUTR, upstream or downstream regions, and intronic variants located more than five base pairs away from exon-intron junctions were also discarded.
-
3.
Filtering by clinical implication. We selected variants of the category assigned as Neurologic in the Clinical Genomic Database (https://research.nhgri.nih.gov/CGD/). This filter was not used with the files obtained from the virtual panel filtering.
-
4.
Filtering by mode of inheritance. trio-WES analysis allows inferring the inheritance pattern of a variant from parents to the proband. We selected variants whose mode of inheritance was compatible with the absence of ID in both parents and the pattern of inheritance of the altered gene, such as heterozygous DNVs in dominant or XL genes, homozygous or compound heterozygous variants in recessive genes, or maternally inherited hemizygous variants in males affecting genes on the X chromosome. Considering that both parents were unaffected and no family history was reported, we decided to exclude the AD inheritance model in our cohort, assuming complete penetrance and non-variable expression.
After variant filtering, we prioritized those variants predicted as Conflicting interpretations of pathogenicity, Uncertain significance (UV), Likely pathogenic (LP), and Pathogenic (P) in ClinVar, InterVar, and VarSome (https://varsome.com/). We discarded those classified as Benign or Likely benign by these databases. Clinical significance of the variants was finally interpreted according to the American College of Medical Genetics and Genomics (ACMG) [35].
Validation of identified variants
Low confidence DNA variants were confirmed by Sanger sequencing. After amplifying DNA of the proband and respective parents, PCR products were sequenced using BigDye Terminator v3.1 (Applied Biosystems, Foster City, CA, USA) following manufacturer’s instructions, and subsequently analyzed on an ABI 3730xl DNA Analyzer (Applied Biosystems). 16p11.2 and 22q13.31q13.33 deletions were confirmed by Cytoscan HD array (Thermo Fisher Scientific, Waltham, MA USA).
Results
Patients description
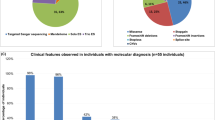
The average age of patients at diagnosis was 29.9 years (Max: 63–Min: 4). 137 (53.94%) of the patients included in this study were male, and 117 (46.06%) were female. The most prevalent comorbidities associated with ID in our cohort were (Supplementary Table S2): epilepsy (71 patients; 27.95%), dysmorphic facial features (30 patients; 11.81%), attention deficit hyperactivity disorder (24 patients; 9.45%), ASD (23 patients; 9.06%), skeletal disorder (21 patients; 8.27%), encephalopathy (15 patients; 5.91%), microcephaly (13 patients; 5.12%), language disorder (12 patients; 4.72%), and psychotic disorder (11 patients; 4.33%).
Sequencing and variant filtering
Trio-based WES was performed to provide a molecular diagnosis. An average read depth of 98X and a sequence coverage ≥30X of 87.4% were obtained. Variant filtering by RefSeq’s coding positions allowed us to reduce the average of variants per exome from 136,522 (Max: 179,475 – Min: 122,954) up to 24,722 (Max: 25,790 – Min: 22,312). After completing the variant filtering pipeline, we selected 117 candidate variants in 90 genes: 70 missense with high-functional impact prediction (59.83%), 18 frameshift (15.38%), 17 nonsense (14.53%), 6 affecting to splice sites (+/- 5 bp; 5.13%), 4 non-frameshift (3.42%), and 2 gross deletions (1.71%) (Tables 1 and 2).
We recognized candidate variants in no more than two unrelated patients in ADNP, ANKRD11, ASXL3, GRIA3, KCNB1, KDM5C, NEXMIF, POLA1, SCN1A, SETBP1, and TCF4 genes. Regarding the latter, we have detected the same heterozygous DNV, NM_001243226.2(TCF4):c.2045 G > A p.(Arg682Gln) in both patients. Such variant has been previously reported as P in ClinVar (ID: 7371). Deleterious variants in TCF4 have been associated with the AD inherited Pitt-Hopkins syndrome (MIM: 610954). Furthermore, we also identified variants of interest in two siblings for LINS and ACSL4 genes.
Suspicion of loss of genetic material in patients 028 P and 086 P (Supplementary Fig. S1) were confirmed by Cytoscan HD array, which identified the de novo heterozygous deletions arr[GRCh38] 16p11.2(29568700_30166678)x1 in 028 P and arr[GRCh38] 22q13.31q13.33(45805040_50759410)x1 in 086 P.
Candidate variants
In total, 97 out of the 254 analyzed patients harbored variants of interest (Supplementary Table S2 and Fig. S2). 73 (62.39%) out of these 117 identified variants were classified as P/LP (Table 1), and the remaining were classified as UV (Table 2). Moreover, 64 (54.7%) out of the 117 variants have not been previously reported. Noteworthy, most selected variants occurred de novo in our cohort. Thus, of the 72 DNVs in our cohort, 52 (72.22%) have been identified in 52 patients who have reached a definitive diagnosis (missense, 34.62%; frameshift, 26.92%; nonsense, 25.0%; non-frameshift, 5.77%; splice site disruption variants, 3.85%; and deletion, 3.85%). The remaining 20 DNVs (27.78%), identified in 19 patients, are missense (80.0%), splice site disruption variants (15.0%), and frameshift (5.0%).
After interpretation and classification of variants, a molecular diagnosis was achieved in 64 patients (25.2%). Thus, 42 of them were diagnosed with AD (65.63%), 13 with AR (20.31%), and 9 with XL (14.06%) forms of ID.
Discussion
Over the past few decades, many efforts have been made to decipher the underlying genetic causes of ID. The advent of NGS has increased both the number of patients in whom a definitive diagnosis of ID has finally been reached and the causative genes associated with this genetically heterogenous neurodevelopmental disorder [27, 36, 37]. In the present study, WES analysis performed in a cohort of 244 ID trios identified P/LP variants in 64 patients, which yields a diagnostic rate of 25.2%. Most affected genes were altered in no more than two probands, which explains the absence of large groups of patients sharing a recurrent clinical diagnosis and corroborates the great locus heterogeneity of ID in our cohort (Table 1).
It is worth noticing the numerous DNVs (72; 61.54%) identified in our cohort, most of them associated with AD/XL inheritance. Other authors have previously reported high ratios of such type of variant, and most of these DNVs have been classified as P [17, 28, 38]. According to this, a high rate of P/LP DNVs (52; 72.22%) was found in our trios. Dominant P DNVs have been related to non-inherited severe ID [27, 39]. Therefore, patients harboring such type of variant could be easier to diagnose than those showing more common forms of inheritance [27]. Thus, likely gene-disrupting DNVs account for ~6–9% of all neurodevelopmental disorders diagnoses [40]. Furthermore, it has been suggested that DNVs in known ID genes could act as modifiers increasing the severity of the primary phenotype. In opposition to likely gene-disrupting DNVs and CNVs, missense DNVs tend to underlie less severe disease forms. Nevertheless, finding a novel DNV in a well-known ID gene does not provide sufficient evidence per se to establish an accurate genotype-phenotype correlation that implicates such variant as causal. For this reason, DNVs should be validated by further functional studies to determine their possible involvement in the phenotype.
Trio-based WES has become essential for the rapid identification of DNVs. In contrast to the singleton approach, trio-based WES provides genotypic information from the parents, allowing precise and immediate discrimination of the de novo origin of a variant [16, 41, 42]. Thus, in trios with unaffected parents and no family history, most of the potentially P variants were de novo [17]. Conversely, in trios with parents sharing a similar altered phenotype, most variants are inherited and have a low probability of being causal. Early application of WES would decrease in healthcare costs since the rapid diagnosis reached in an appreciable number of patients would end their long diagnostic odyssey and no further genetic testing would be necessary [14, 33]. Most patients from our cohort are adults, indicating that they have never been genetically tested or have undergone many inefficient genetic tests during their lifetime. According to other studies, WES can be postulated as a powerful tool to reach a definitive diagnosis in adults [18, 19, 21]. So, WES has allowed us to obtain a diagnosis of Kabuki Syndrome 2 (MIM: 300867) in patient 002 P (early 40 s), Bainbridge-Ropers syndrome (MIM: 615485) in 217 P (early 50 s), or Kleefstra syndrome 2 (MIM: 617768) in 215 P (late 50 s). Nevertheless, we also achieved the diagnosis in younger patients, such as Kabuki syndrome 1 (MIM: 147920) in patient 030 P (early 10 s) or Rett syndrome (MIM: 613454) in 161 P (early 10 s). Regarding this, it should be noted that a detailed clinical characterization of the patient and a complete family history could be essential to determine a possible genotype-phenotype correlation or to elucidate between the possible differential diagnosis in complex patients [8, 13, 43]. Nonetheless, in patients with less complex phenotypes who might be easier to diagnose, the cost-effectiveness of WES seems to fall below that of gene panel analysis [32].
The slightly higher percentage of males (53.94%) in our cohort could be partially explained by the fact that males are more susceptible to XL ID [44]. However, out of nine patients harboring P/LP XL variants, seven are females (Table 1). Six of them were diagnosed with dominant XL disorders. The remaining one (091 P) was diagnosed with a recessive XL disorder (Menkes disease; MIM: 3094009), regardless of having a unique heterozygous frameshift variant in ATP7A gene. Although there is a chance that a second mutational event in this gene has evaded detection by WES in this patient [13], or that an X-chromosome skewed inactivation may be involved [43], it has been reported that female carriers may have a variable presence and/or severity of the clinical features associated with such disorder [45]. Furthermore, we also have identified 13 patients harboring P/LP variants in AR ID genes. Nine out of these 13 patients were compound heterozygotes for genes associated with ID, and the remaining four were homozygous (Table 1). According to Fitzgerald et al., most patients with AR ID in our cohort were syndromic [46].
An interesting point of our study was the concurrent identification of P/LP variants in two independent genes in the same patient (097 P, 154 P and 228 P). In patient 097 P, both affected genes were associated with similar phenotypes, greatly hindering the diagnostic process. Thus, the phenotype of patient 097 P, showing ID, epilepsy, disruptive behavior, dysmorphic facial features, brachycephaly, bilateral transverse palmar crease, multiple angiomas, obesity, and altered vision, could be associated either with the nonsense variant identified in ADNP (AD inherited Helsmoortel-van der Aa syndrome; MIM: 615873) or with the missense in PHIP (AD inherited Chung-Jansen syndrome; MIM: 6179919). A better and more exhaustive clinical history could help us to establish a correct genotype-phenotype relationship. Nonetheless, we should consider that the resulting phenotype in these patients could be due to the overlap of the two syndromes [47]. Therefore, further functional studies would be required to better understand the potential effect of these variants over protein functionality to successfully discard the non-causal variant and finally reach a definitive diagnosis. Equally, such co-occurrence of variants of interest in two independent genes has also been identified in patients harboring UV variants in our cohort (104 P, 108 P, 133 P, 136 P, and 209 P). This fact further complicates the identification of the true causal variant in these patients.
Another remarkable finding was that genetic diagnosis had not been reached in 157 patients (61.81%). Nevertheless, a comparison between diagnosed and undiagnosed patients classified according to their ID grade showed similar percentages for each category in both groups. Therefore, a non-diagnostic WES result does not exclude the potential causal variant within the exome dataset. In this regard, it should be noted that reported variants have been detected by both the virtual panel and WES in well-known ID genes. However, the exclusion of variants only detected by WES in genes not well characterized or having limited available information at the time of this report could be considered a limitation of this study. Thus, systematic reanalysis of non-diagnostic WES data could lead to an improved yielding in the identification of P/LP variants and, consequently, to an increase in diagnostic rate [16, 17, 31, 41]. Likewise, those variants classified as UV could also benefit from successive exome reanalysis since new clinical interpretation based on the acquired knowledge could reclassify them as either pathogenic or benign [31]. In this regard, our study identified UV in 33 (12.99%) patients. Finding an UV in a genetic test can cause frustration, anxiety, and uncertainty for patients and their families, so managing these cases requires careful pre- and post-test genetic counseling [15].
If WES remains non-diagnostic after reanalysis, other diagnostic tools are still available to achieve that objective, including karyotyping to detect balanced chromosomal rearrangements, chromosome microarray analysis, or mitochondrial DNA sequencing [31]. Moreover, there is a current trend for calling CNVs from exome because their presence could be more than likely and their identification could lead to a diagnostic yield increasing [48]. Even though our study does not include information on CNVs, the presence of apparent homozygous variants inherited from only one parent in chromosome 16p of patient 028 P and 22q of 086 P raised suspicion of hemizygous deletions affecting these regions, which were finally confirmed by Cytoscan HD (Supplementary Fig. S1). The de novo deletions in 16p11.2 (MIM: 611913) and 22q13.31q13.33 (MIM: 606232) were previously associated with ID and ASD, and Phelan-McDermid syndrome, respectively [49, 50]. Therefore, further studies are needed to determine the ratio of CNVs present in our cohort, particularly in the subset of undiagnosed patients without array CGH analysis. Another tool, Whole Genome Sequencing (WGS), could have even better diagnostic yielding than WES due to its ability for sequencing non-coding regions and its greater sensitivity to detect CNVs [12, 40]. Nevertheless, its high cost of sequencing, and the arduous complexity in analyzing the data generated, hamper that WGS could be considered the diagnostic tool of first choice [12]. Finally, epigenetic changes and polygenic inheritance are other possible causes that should be considered for ID diagnosis [7, 14]. It is challenging to know the proportion of monogenic or polygenic components of a genetic condition in a specific cohort [17]. Identifying disease-associated genes by NGS has allowed achieving an increasing number of novel diagnoses, most of them associated with dominant disorders. However, polygenic inheritance may likely hide very rare recessive disorders that could be described in the near future.
In summary, trio-WES analysis has proven to be an essential tool for the genetic diagnosis of ID in our cohort, yielding a diagnostic rate of 25.2%. Moreover, identifying 31 novel P/LP variants in 27 patients expanded the molecular spectrum associated with ID. On the other hand, reanalysis of WES seems to be the best way to achieve a definitive genetic diagnosis either in patients with no test results or classified as of uncertain significance. Continuous updating of the information contained in the main consulted databases, development of novel bioinformatics algorithms of analysis, as well as a detailed clinical history, result mandatory for the precise identification and interpretation of variants. Therefore, the collaboration between clinicians, scientists, patients, and families becomes essential to provide further insight into the underlying genetic causes of ID.
Data availability
Identified variants have been deposited on the Leiden OpenVariation Database (LOVD, https://www.lovd.nl/). Complementary data that support the findings of this study are available from the corresponding author upon reasonable request.
References
Patel DR, Cabral MD, Ho A, Merrick J. A clinical primer on intellectual disability. Transl Pediatr. 2020;9:S23–35.
Ropers HH. Genetics of early onset cognitive impairment. Annu Rev Genomics Hum Genet. 2010;11:161–87.
Casanova EL, Sharp JL, Chakraborty H, Sumi NS, Casanova MF. Genes with high penetrance for syndromic and non-syndromic autism typically function within the nucleus and regulate gene expression. Mol Autism. 2016;7:18.
Darvish H, Azcona LJ, Tafakhori A, Mesias R, Ahmadifard A, Sanchez E, et al. Phenotypic and genotypic characterization of families with complex intellectual disability identified pathogenic genetic variations in known and novel disease genes. Sci Rep. 2020;10:968.
Ellison JW, Rosenfeld JA, Shaffer LG. Genetic basis of intellectual disability. Annu Rev Med. 2013;64:441–50.
Jamra R. Genetics of autosomal recessive intellectual disability. medizinische Genet. 2018;30:323–7.
Wieczorek D. Autosomal dominant intellectual disability. Medizinische Genet. 2018;30:318–22.
Neri G, Schwartz CE, Lubs HA, Stevenson RE. X-linked intellectual disability update 2017. Am J Med Genet Part A. 2018;176:1375–88.
Musante L, Ropers HH. Genetics of recessive cognitive disorders. Trends Genet. 2014;30:32–9.
Chiurazzi P, Pirozzi F. Advances in understanding—genetic basis of intellectual disability. F1000Research. 2016;5:599.
Ilyas M, Mir A, Efthymiou S, Houlden H. The genetics of intellectual disability: advancing technology and gene editing. F1000Research. 2020;9:22.
Harripaul R, Noor A, Ayub M, Vincent JB. The use of next-generation sequencing for research and diagnostics for intellectual disability. Cold Spring Harb Perspect Med. 2017;7:a026864.
Amor DJ. Investigating the child with intellectual disability. J Paediatr Child Health. 2018;54:1154–8.
Bass N, Skuse D. Genetic testing in children and adolescents with intellectual disability. Curr Opin Psychiatry. 2018;31:490–5.
Vickers RR, Gibson JS. A review of the genomic analysis of children presenting with developmental delay/intellectual disability and associated dysmorphic features. Cureus. 2019;11:e3873.
Bowling KM, Thompson ML, Amaral MD, Finnila CR, Hiatt SM, Engel KL, et al. Genomic diagnosis for children with intellectual disability and/or developmental delay. Genome Med. 2017;9:43.
Wright CF, McRae JF, Clayton S, Gallone G, Aitken S, FitzGerald TW, et al. Making new genetic diagnoses with old data: iterative reanalysis and reporting from genome-wide data in 1,133 families with developmental disorders. Genet Med. 2018;20:1216–23.
Sabo A, Murdock D, Dugan S, Meng Q, Gingras M-C, Hu J, et al. Community‐based recruitment and exome sequencing indicates high diagnostic yield in adults with intellectual disability. Mol Genet Genom Med. 2020;8:e1439.
Benson KA, White M, Allen NM, Byrne S, Carton R, Comerford E, et al. A comparison of genomic diagnostics in adults and children with epilepsy and comorbid intellectual disability. Eur J Hum Genet. 2020;28:1066–77.
Topper S, Ober C, Das S. Exome sequencing and the genetics of intellectual disability. Clin Genet. 2011;80:117–26.
Snoeijen-Schouwenaars FM, van Ool JS, Verhoeven JS, van Mierlo P, Braakman HMH, Smeets EE, et al. Diagnostic exome sequencing in 100 consecutive patients with both epilepsy and intellectual disability. Epilepsia 2019;60:155–64.
Smaili W, Elalaoui SC, Zrhidri A, Raymond L, Egéa G, Taoudi M, et al. Exome sequencing revealed a novel homozygous METTL23 gene mutation leading to familial mild intellectual disability with dysmorphic features. Eur J Med Genet. 2020;63:103951.
Banihashemi S, Tahmasebi-Birgani M, Mohammadiasl J, Hajjari M. Whole exome sequencing identified a novel nonsense INPP4A mutation in a family with intellectual disability. Eur J Med Genet. 2020;63:103846.
Palumbo P, Di Muro E, Accadia M, Benvenuto M, Di Giacomo MC, Castellana S, et al. Whole exome sequencing reveals a novel AUTS2 in-frame deletion in a boy with global developmental delay, absent speech, dysmorphic features, and cerebral anomalies. Genes. 2021;12:229.
Martínez F, Caro-Llopis A, Roselló M, Oltra S, Mayo S, Monfort S, et al. High diagnostic yield of syndromic intellectual disability by targeted next-generation sequencing. J Med Genet. 2017;54:87–92.
Pekeles H, Accogli A, Boudrahem-Addour N, Russell L, Parente F, Srour M. Diagnostic yield of intellectual disability gene panels. Pediatr Neurol. 2019;92:32–6.
Stojanovic JR, Miletic A, Peterlin B, Maver A, Mijovic M, Borlja N, et al. Diagnostic and clinical utility of clinical exome sequencing in children with moderate and severe global developmental delay / intellectual disability. J Child Neurol. 2020;35:116–31.
de Ligt J, Willemsen MH, van Bon BWM, Kleefstra T, Yntema HG, Kroes T, et al. Diagnostic exome sequencing in persons with severe intellectual disability. N Engl J Med. 2012;367:1921–9.
Farwell KD, Shahmirzadi L, El-Khechen D, Powis Z, Chao EC, Tippin Davis B, et al. Enhanced utility of family-centered diagnostic exome sequencing with inheritance model–based analysis: results from 500 unselected families with undiagnosed genetic conditions. Genet Med. 2015;17:578–86.
Carneiro T, Krepischi A, Souza da Costa S, Tojal da Silva I, Vianna-Morgante A, Valieris R, et al. Utility of trio-based exome sequencing in the elucidation of the genetic basis of isolated syndromic intellectual disability: illustrative cases. Appl Clin Genet. 2018;11:93–8.
Srivastava S, Love-Nichols JA, Dies KA, Ledbetter DH, Martin CL, Chung WK, et al. Meta-analysis and multidisciplinary consensus statement: exome sequencing is a first-tier clinical diagnostic test for individuals with neurodevelopmental disorders. Genet Med. 2019;21:2413–21.
LELM Vissers, Van Nimwegen KJM, Schieving JH, Kamsteeg EJ, Kleefstra T, Yntema HG, et al. A clinical utility study of exome sequencing versus conventional genetic testing in pediatric neurology. Genet Med. 2017;19:1055–63.
Vrijenhoek T, Middelburg EM, Monroe GR, van Gassen KLI, Geenen JW, Hövels AM, et al. Whole-exome sequencing in intellectual disability; cost before and after a diagnosis. Eur J Hum Genet. 2018;26:1566–71.
Satterstrom FK, Kosmicki JA, Wang J, Breen MS, De Rubeis S, An J-Y, et al. Large-scale exome sequencing study implicates both developmental and functional changes in the neurobiology of Autism. Cell. 2020;180:568–584. e23
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–23.
Di Resta C, Pipitone G, Carrera P, Ferrari M. Current scenario of the genetic testing for rare neurological disorders exploiting next generation sequencing. Neural Regen Res. 2021;16:475–81.
Fell CW, Nagy V. Cellular models and high-throughput screening for genetic causality of intellectual disability. Trends Mol Med. 2021;27:220–30.
The Deciphering Developmental Disorders Study. Prevalence and architecture of de novo mutations in developmental disorders. Nature 2017;542:433–8.
Reichenberg A, Cederlöf M, McMillan A, Trzaskowski M, Kapra O, Fruchter E, et al. Discontinuity in the genetic and environmental causes of the intellectual disability spectrum. Proc Natl Acad Sci. 2016;113:1098–103.
Wilfert AB, Sulovari A, Turner TN, Coe BP, Eichler EE. Recurrent de novo mutations in neurodevelopmental disorders: properties and clinical implications. Genome Med. 2017;9:101.
Wenger AM, Guturu H, Bernstein JA, Bejerano G. Systematic reanalysis of clinical exome data yields additional diagnoses: implications for providers. Genet Med. 2017;19:209–14.
Wang J, Wang Y, Wang L, Chen WY, Sheng M. The diagnostic yield of intellectual disability: combined whole genome low-coverage sequencing and medical exome sequencing. BMC Med Genomics. 2020;13:70.
De Luca C, Race V, Keldermans L, Bauters M, Van Esch H. Challenges in molecular diagnosis of X-linked Intellectual disability. Br Med Bull. 2020;133:36–48.
Gao C, Wang X, Mei S, Li D, Duan J, Zhang P, et al. Diagnostic yields of trio-WES accompanied by CNVseq for rare neurodevelopmental disorders. Front Genet. 2019;10:485.
Smpokou P, Samanta M, Berry GT, Hecht L, Engle EC, Lichter-Konecki U. Menkes disease in affected females: The clinical disease spectrum. Am J Med Genet Part A. 2015;167:417–20.
Fitzgerald TW. Large-scale discovery of novel genetic causes of developmental disorders. Nature 2015;519:223–8.
Posey JE, Harel T, Liu P, Rosenfeld JA, James RA, Coban Akdemir ZH, et al. Resolution of disease phenotypes resulting from multilocus genomic variation. N Engl J Med. 2017;376:21–31.
Gilissen C, Hehir-Kwa JY, Thung DT, van de Vorst M, van Bon BWM, Willemsen MH, et al. Genome sequencing identifies major causes of severe intellectual disability. Nature. 2014;511:344–7.
Shinawi M, Liu P, Kang SHL, Shen J, Belmont JW, Scott DA, et al. Recurrent reciprocal 16p11.2 rearrangements associated with global developmental delay, behavioural problems, dysmorphism, epilepsy, and abnormal head size. J Med Genet. 2010;47:332–41.
Phelan K, McDermid HE. The 22q13.3 deletion syndrome (Phelan-McDermid syndrome). Mol Syndromol. 2011;2:186–201.
Acknowledgements
We are sincerely grateful to patients and their families for participating in this study. This study has been funded by Instituto de Salud Carlos III through the project PI19/00809 (Co-funded by European Regional Development Fund/European Social Fund “A way to make Europe”/“Investing in your future”). We also acknowledge support from Fundación María José Jove.
Author information
Authors and Affiliations
Contributions
AJBF and MAB should be considered joint first author. Study concept and design, AJBF, MAB, JB, AC; writing – original draft preparation, AJBF, MAB; writing – review and editing, JB, AC, AJBF, MAB; clinical diagnosis or clinical data gathering, MTF, MFP, PC; bioinformatic analysis, JA, AJBF, SDR; genetic data acquisition or analysis, AJBF, MAB, FB, SDR. All authors have approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
The study was conducted according to the guidelines of the Declaration of Helsinki, and approved by the Institutional Review Board of the Research Ethics Committee of Galicia (Comité Ético de Investigación Galicia; the only IEC authorized in this autonomous region); Number: 2015/608; Approval: 18-November-2015; Title: DISECMAS: Bases genéticas de la discapacidad intelectual: aplicación de las nuevas tecnologías de secuenciación masiva al análisis de variantes genéticas. Informed consent was obtained from all participants included in the study.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Brea-Fernández, A.J., Álvarez-Barona, M., Amigo, J. et al. Trio-based exome sequencing reveals a high rate of the de novo variants in intellectual disability. Eur J Hum Genet 30, 938–945 (2022). https://doi.org/10.1038/s41431-022-01087-w
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-022-01087-w
This article is cited by
-
Exome sequencing improves the molecular diagnostics of paediatric unexplained neurodevelopmental disorders
Orphanet Journal of Rare Diseases (2024)
-
Exome sequencing—one test to rule them all?
European Journal of Human Genetics (2022)
