
Abstract
The diagnostic and clinical benefits of genomic sequencing are being increasingly demonstrated across multiple rare genetic conditions. Despite the expanding clinical literature, there is a significant paucity of health economics evidence to inform the prioritization and implementation of genomic sequencing. This study aims to evaluate whether genomic sequencing for pediatric-onset mitochondrial disorders (MDs) is cost-effective and cost-beneficial relative to conventional care from an Australian healthcare system perspective. Two independent and complementary health economic modeling approaches were used. Approach 1 used a decision tree to model the costs and outcomes associated with genomic sequencing and conventional care. Approach 2 used a discrete-event simulation to incorporate heterogeneity in the condition and clinical practice. Deterministic and probabilistic sensitivity analyses were performed. Genomic sequencing was less costly and more effective compared with conventional care, saving AU$1997 (Approach 1) to AU$8823 (Approach 2) per child tested, while leading to an additional 11 (Approach 1) to 14 (Approach 2) definitive diagnoses per 100 children tested. The mean monetary value of the incremental benefits of genomic sequencing was estimated at AU$5890 (95% CI: AU$5730−$6046). Implementation of genomic sequencing for MDs in Australia could translate to an annual cost-saving of up to AU$0.7 million. Genomic sequencing is cost-saving relative to traditional investigative approaches, while enabling more diagnoses to be made in a timely manner, offering substantial personal benefits to children and their families. Our findings support the prioritization of genomic sequencing for children with MDs.
Similar content being viewed by others
Introduction
Mitochondrial disorders (MDs) are rare genetic conditions, evidenced in more than 1 per 5000 live births [1, 2], caused by the variants in mitochondrial (mtDNA) or nuclear DNA (nDNA) affecting mitochondrial oxidative phosphorylation. The clinical manifestations of MDs are greatly heterogeneous, commonly involving multiple organs, and particularly those that are highly dependent on aerobic metabolism [3]. Due to their diverse phenotypes, MDs are associated with a complex and burdensome diagnostic odyssey, often involving extensive investigations, including biochemical testing, neuroimaging, biopsies, histology, and enzymology, while requiring multiple specialist consultations that often result in conflicting diagnoses [4,5,6]. MDs have detrimental effects on patient and carer quality of life [7].
Genomic sequencing has provided an opportunity to reshape the diagnostic approach to genetic conditions, and in many countries with advanced economies, genomic sequencing has started to be integrated into mainstream clinical care [8]. A growing body of evidence demonstrates the diagnostic, clinical, and personal outcomes generated by genomic sequencing [9,10,11]. The health economics evidence-base, however, is limited [12], even though evidence of cost-effectiveness is required to support reimbursement decisions and clinical guidelines by national health technology assessment (HTA) bodies. In the context of MDs, genomic sequencing provides a definitive diagnosis in 35−70% of patients [13], potentially enabling a range of outcomes that are highly valued by society [14, 15], such as knowledge about the cause of the condition, improvements in the process and outcomes of medical care, ending diagnostic odyssey and associated uncertainty, and facilitating access to peer support or clinical trials, which could justify the additional cost of sequencing. To date, there have been no studies evaluating the cost-effectiveness of genomic sequencing specifically for patients suspected with MDs. Access to genomic sequencing is, therefore, fragmented and can only be gained through clinical research studies, potentially through hospital budgets (in some countries like Australia), or privately, which affects equity and economic healthcare system objectives.
Our study aims to evaluate whether genomic sequencing in children presenting with clinical indications of MDs is cost-effective and cost-beneficial relative to the conventional diagnostic pathway from an Australian healthcare system perspective. Utilizing primary clinical and economic data collected prospectively as part of the Australian Genomics Mitochondrial Disease clinical project and retrospectively through the Sydney Children’s Hospitals Network registry, we explored two independent and complementary health economic modeling approaches that generate the required health economics evidence for the prioritization of genomic sequencing for children with MDs.
Methods
Study design and participants
This study was informed by data from the Australian Genomics Health Alliance Mitochondrial Disease clinical project (AGHA cohort) and a historical cohort of patients with MDs from the Sydney Children’s Hospitals Network (historical SCHN cohort). The AGHA cohort consisted of 78 pediatric-onset patients (age of onset ≤ 16 years) with suspected MDs prospectively recruited for genomic sequencing between March 2017 and July 2019. For these individuals, nuclear genomic sequencing was initially performed, and if this was nondiagnostic, they went on to have mtDNA genomic sequencing. Inclusion criteria were the diagnosis of “probable MD”, defined as a score > 4 using a modified Nijmegen scoring system [16], as described in the online appendices (Table S1). Exclusion criteria were an existing molecular diagnosis or a Nijmegen score of 4 or less, as determined by an intake review committee.
The historical SCHN cohort included 61 pediatric patients referred to the metabolic clinic at SCHN between Mar 2001 and Mar 2018 who were suspected of having a MD based on clinical, biochemical, imaging, and histological grounds. These patients had undertaken traditional diagnostic workup, including metabolic, imaging and histopathological investigations, and genetic testing. The Nijmegen scores for this cohort at first and last clinical presentation to the metabolic or neurology clinic were assessed retrospectively through a review of medical files by two members of the research team (SB and RR) to ensure consistency between cohorts. Although clinicians are increasingly using genomic sequencing, these traditional diagnostic workups are considered as the conventional practice, given that genomic sequencing for MDs is not federally funded in Australia.
Ethical approval was granted from the Melbourne Health Human Research Ethics Committee (MH-HREC reference number HREC/16/MH/251) and individual patient consent was received from all patients recruited prospectively. Ethical approval was also granted from the Sydney Children’s Hospitals Network Human Research Ethics Committee (SCHN-HREC reference number HREC/18/SCHN/362) for accessing retrospective registry patient data. For this, the need for individual patient consent was waived and data were de-identified.
Economic evaluation
Two independent and complementary approaches were used to model the costs and outcomes associated with genomic sequencing and conventional diagnostic care in children with probable MDs:
-
Approach 1 utilized a decision tree that provided a standardized description of the diagnostic pathways of the different comparators. The modeled pathways were conceptualized through a mapping exercise of clinical practice across different states in Australia in collaboration with participating centers in the AGHA study aiming to reach clinical consensus. Resource use associated with the clinical utility of genomic sequencing was identified through a clinician survey for each of the children participating in the AGHA national cohort.
-
Approach 2 utilized a discrete-event simulation model that incorporated patient-level data from the historical SCHN cohort and the patients from the AGHA cohort who had been referred from SCHN. The model was developed to reflect key events in children’s clinical pathways, which enabled an incorporation of condition heterogeneity and heterogeneity in clinical practice. The analysis was restricted to the SCHN patients to ensure consistency in the costing process.
In line with best practice recommendations [17], to ensure face validity for each modeling approach, model structures, associated assumptions, and model inputs were all approved by leading clinicians both internal and external to the research team. An internal validation of the models was also performed by manual calculation of rhe expected values.
Model structures
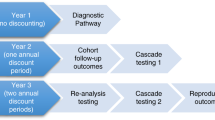
Approach 1a compared initially three diagnostic strategies: (a) genomic sequencing, (b) late genomic sequencing, and (c) conventional diagnostic pathway (Fig. 1). Genomic sequencing options involved either exome sequencing (ES) followed by mtDNA sequencing (herein labeled as “ES ± mtDNA”), if exome sequencing did not result in a definitive diagnosis, or genome sequencing (GS). Patients were regarded as diagnosed if they had pathogenic or likely pathogenic variants, defined by standard criteria [18], in a gene consistent with their presentation. Given that genome sequencing could offer the advantage of identifying clinically relevant noncoding pathogenic variants and improved detection of copy number variants compared with ES ± mtDNA, GS and ES ± mtDNA are modeled separately in an additional analysis (Approach 1b).

Notes: MRI/MRS magnetic resonance imaging/magnetic resonance spectroscopy; MRC mitochondrial respiratory chain; Dx diagnosis; Mx management; [+] plus sign denotes similar model structure following genomic sequencing as the one presented in the genomic sequencing pathway.
Across all strategies, children with a probable MD initially have specific biochemistry tests and magnetic resonance imaging (MRI)/magnetic resonance spectroscopy (MRS) (Tier 1). Following Tier 1 in the genomic sequencing pathway, children go straight to singleton genomic sequencing. Depending on the identified variants, segregation testing of family members was performed to confirm or clarify the genetic diagnosis. Following genomic sequencing, children may have changes in their clinical management (e.g., changes in treatment or surveillance). Following Tier 1 in the conventional diagnostic pathway, children generally undergo biopsies, histology and enzymology (Tier 2), and genetic testing (Tier 3). In the late genomic sequencing pathway, children will follow the conventional diagnostic pathway and receive genomic sequencing after Tier 2 testing (Fig. 1).
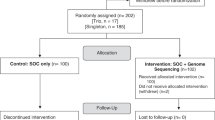
The modeled pathways in Approach 2 are shown in Fig. 2. Following the first metabolic or neurology clinic consultation in the conventional pathway, children with a probable MD could receive a presumed diagnosis either through genetic testing or through clinical and biochemical studies. In this analysis, we allowed age to vary within each simulation to reflect the age distribution observed in the SCHN data. Following a presumed diagnosis through clinical and biochemical studies, the majority of children (92%) still proceeded to genetic testing for a confirmed molecular diagnosis. In the genomic sequencing arm, children go directly to genomic sequencing after the initial metabolic or neurology clinic consultation. In the absence of a confirmed genetic or genomic diagnosis, diagnostic investigations will continue. In the presence of a confirmed diagnosis, some investigations and specialist appointments may still be required.

Notes: Dx diagnosis; Ix investigations.
Cost parameters
Unit costs included in Approach 1 were predominantly sourced from the Australian Medicare Benefits Schedule (MBS) [19] and the Victorian Clinical Genetics Services (VCGS) price list (available from vcgs@vcgs.org.au), and are listed in Table S2. Given the age of the cohort and severity of MDs, the mean cost of biopsy (AU$5839), including surgeon and theater costs, as well as ward medical and nursing costs, was estimated using micro-costing methods based on the patient-level cost data of 24 children in the AGHA and historical SCHN cohorts who had biopsies. The costs were obtained from the Royal Children’s Hospital Melbourne and the SCHN costing departments. To estimate the cost of genetic testing in the conventional diagnostic pathway, a review of medical records in the historical SCHN cohort was conducted by a member of the research team (SB) to identify the number and type of genetic tests performed for each child. Unit cost estimates from VCGS were then applied to each genetic test identified (Table S2). The costs of genomic sequencing pathways included the cost of genomic sequencing, segregation tests, genetics consultations, and resource use associated with the test’s clinical utility (Table S2). A cost of $4150 was used for genome sequencing, which included analysis for 101−400 genes and whole mtDNA genome analysis. For exome sequencing (analysis for 201−400 genes) and mtDNA sequencing, a cost of $2400 and $1150, respectively, was used. To cost resource use associated with changes in clinical management following testing, namely resources associated with the test’s clinical utility, a purpose-designed survey was administered to all participating clinicians in the AGHA cohort. The survey, which is available in the online supplemental material, asked information about treatment changes and changes in screening tests and subspecialist referrals following the genomic sequencing results. Clinical utility-related costs following a definitive or non-definitive diagnosis were assumed to be similar across all comparators. Given that secondary findings from genomic sequencing (i.e., results unrelated to the test indication) were not explored as part of the clinical studies, and that the return of these findings would relate to a separate reimbursement decision, at least within the Australian context, healthcare resource use and outcome implications from the return of secondary findings were not modeled.
For Approach 2, direct hospital micro costing data for inpatient and outpatient encounters were obtained from the historical (n = 42) and AGHA (n = 24) SCHN cohorts (Table S4). Cost of diagnostic workup included in the analysis consisted of four components: (1) cost of pathology and imaging testing for diagnostic purposes of MDs (e.g., biochemistry, hematology, histopathology, cytogenetics, ultrasound, MRI/MRS, and CT), (2) cost of procedures related to diagnostic investigations (e.g., general anesthesia for MRI/MRS and CT and surgery cost for biopsy), (3) cost of hospital stay for diagnostic purposes, and (4) cost of outpatient specialist consultation for diagnostic purposes. These costs were then apportioned to the relevant time periods as shown in Fig. 2 and Table S4. Approaches 1 and 2 used the same costs for genetic testing and genomic sequencing.
Probability and other parameters
The diagnostic yield (0.38) of singleton genomic sequencing, followed by segregation testing when necessary, and the proportion of participants who had segregation testing and the number of segregation tests performed per child were sourced from the AGHA cohort (Table S3). The probability of having clinical management initiated or canceled following a definitive diagnosis was 0.21 and 0.11, respectively. Following a non-definitive diagnosis, the probability of having clinical management initiated or canceled was 0.17 and 0.13, respectively. To standardize the diagnostic yield in the conventional diagnostic pathway, two members of our research team (JC and SB) identified the number of children in the historical SCHN cohort who received a definitive genetic diagnosis and assessed whether a genomic diagnosis could have been made. The proportion of genetic diagnoses out of the overall genomic diagnoses in the historical SCHN cohort was used to estimate the number of diagnoses that would have been made by conventional diagnostic pathways in the AGHA cohort. Following this process, a diagnostic yield of 0.27 was estimated for the conventional diagnostic pathway. This estimate was also varied by 50% in a sensitivity analysis. Tables S2 and S3 list all probabilities and other parameters used in the two modeling approaches.
Analyses
An expected value analysis was applied to estimate the incremental cost-effectiveness and cost benefit of genomic sequencing relative to conventional diagnostic pathway in children with suspected MDs. Analyses were undertaken from an Australian healthcare system perspective based on the outcomes of cost per additional definitive diagnosis and net benefit, defined as the difference between the incremental monetary value of the benefits generated and the incremental monetary value of the costs. To ensure that analyses captured longer-term resource use implications associated with the clinical utility of genomic sequencing, a time horizon from the first metabolic or neurology clinic consultation until the age of 18 was used. Given that differences in survival probabilities between genomics and nongenomics pathways on the basis of a confirmed molecular diagnosis were not expected, children were assumed to be alive during the modeled time horizon. All costs are in 2020 Australian dollars, and an annual discount rate of 0.05 was used as recommended in Australia [20]. Alternative discounting rates (i.e., 0.015 and 0.035) were explored in a sensitivity analysis. The incremental monetary value of the benefits generated by genomic sequencing relative to conventional care was estimated based on the findings of the economic evaluation and the marginal utilities of our published discrete-choice experiment (DCE) [15], using the compensating variation formula [21], and with the marginal willingness-to-pay for time-related attributes being discounted using the recommended-in-Australia 0.05 annual discounting rate. The process of estimating the welfare gain of genomic sequencing has been extensively described in our published DCE [15].
For a comprehensive evaluation of decision uncertainty related to the model parameters and condition or patient heterogeneity, probabilistic sensitivity analyses were conducted using Monte Carlo simulations. The probabilistic analysis generated 10,000 iterations of the model, whereby each iteration provides a different set of parameter estimates drawn from their corresponding parameter probability distribution (Approaches 1 and 2), with 10,000 children being simulated per alternative within each iteration (Approach 2). Gamma and Beta distributions were used for cost and probability parameters, respectively (Tables S1–S3) [22]. Time parameters used in Approach 2 were assigned a Gamma distribution, apart from the time to access genomic sequencing and the time to genomic sequencing results where a Uniform distribution was used (Table S4). Cost-effectiveness acceptability curves (CEACs) were used to demonstrate decision uncertainty by plotting the probability of each diagnostic option being cost-effective across a range of willingness to pay thresholds per additional diagnosis [23].
Deterministic sensitivity analyses were also conducted, as previously described, to explore how variations in key model inputs impacted on model results. Given potential variations in the cost and diagnostic yield of GS for MDs within Australia and beyond, an additional sensitivity analysis to Approach 1b was performed to demonstrate how the incremental cost-effectiveness ratio (ICER) of GS relative to ES ± mtDNA varies depending upon variations in the cost ($2500−$5000) and incremental yield (0.01−0.22) of GS. Finally, a scenario analysis was additionally performed in Approach 1, given that patients receiving non-definitive diagnosis from genomic sequencing may be offered additional investigations to explore the underlying etiology, particularly in children with strong indication of MDs based on clinical features, metabolic, and imaging studies, or those with novel variant(s) identified in known disease-causing genes. Such investigations, which are mostly conducted in the research laboratories, include biopsy, enzymology, specific biochemistry tests, reanalysis of sequencing data, other molecular tests, and/or functional studies. This scenario analysis included the cost and diagnostic yield of additional investigations following a non-definitive genomic diagnosis in the ES and GS pathways. Nondefinitive diagnosis includes patients with negative results and those with variants of unknown significance (VUS). The relevant resource use and diagnostic rates were informed by the investigations performed and initiated in the AGHA cohort (Table S2) and test results obtained (Table S3). The models were developed and analyzed in TreeAge Pro 2020 software.
Results
The results of the cost-effectiveness analysis for Approaches 1 and 2 are available in Table 1. In Approach 1a, genomic sequencing of all children with suspected MDs resulted in a mean-per-child cost of $6757, with 38 children out of 100 being definitively diagnosed. This cost was attributed to Tier 1 testing (31.6%), genomic sequencing (62.9%), and changes in clinical management (5.5%) for children with definitive ($215) and nondefinitive ($469) molecular diagnosis. The mean-per-child cost for the conventional diagnostic pathway was $8754, with 27 out of 100 children being definitively diagnosed. This cost was attributed to Tier 1 testing (24.4%), Tier 2 testing (62.7%), genetic testing (8.4%), and changes in clinical management (4.6%).
Therefore, genomic sequencing resulted in a cost-saving of $1997 per child tested compared with conventional care. Late application of genomic sequencing was dominated by early genomic sequencing, as it was more costly and less effective. The probability of genomic sequencing being cost-effective was >95% across any threshold of willingness-to-pay per additional definitive diagnosis (Fig. 3). The scenario analysis indicated that the additional investigations conducted following a non-definitive genomic diagnosis increased diagnostic yield from 0.38 to 0.44, reducing therefore the cost per definitive genomic diagnosis by approximately $2000.

Note: The graph plots the probability of each diagnostic alternative being cost-effective across a range of willingness to pay values per additional definitive diagnosis.
In approach 1b, ES ± mtDNA resulted in a mean-per-child cost of $6406 and 33 definitive diagnoses in 100 children tested (Table 1). GS pathway was $764 more costly per child ($7170) relative to ES ± mtDNA and yielded 44 definitive diagnoses. Thus, GS had incremental cost-effectiveness ratio of $6945 per additional definitive diagnosis compared with ES ± mtDNA. Table S5 presents the outcomes of GS and ES ± mtDNA across the eight attributes included in our DCE [15]. The attributes are (1) number of children who receive a genetic diagnosis, (2) chance of improving the process of child’s medical care, (3) availability of treatments, (4) time until the child has the test, (5) time between the test and results, (6) enabling access to other services and professional or peer support, (7) enabling access to clinical trials, and (8) cost of testing. Based on these differences, the mean monetary value of the incremental benefits of GS relative to ES ± mtDNA was estimated at $425 (95% CI: $412−$440), which is equivalent to $3863 per additional diagnosis. As shown in Figure S1, at this threshold of WTP per additional diagnosis, ES ± mtDNA appears to be the most cost-effective option but only with 60% probability of being cost-effective relative to GS. Figure 4 presents how the ICER of GS relative to ES ± mtDNA varies depending on the cost of delivering GS and the incremental diagnostic yield of GS. If the cost of delivering GS is less than $3500, GS is a more effective and less costly diagnostic strategy compared with ES ± mtDNA. At a cost of $4000, the ICER of GS is less than $2000 per additional definitive diagnosis if diagnostic yield is more than 5 percentage points higher than ES ± mtDNA (i.e., ≥ 38%). However, for higher costs of delivering GS, there is more uncertainty as to which of the two genomic sequencing strategies is more cost-effective.

Note: The gray curves below the x-axis are presented for illustrative purposes only, as they represent negative ICERs, to demonstrate the dominance of GS relative to ES ± mtDNA, namely that GS is both more effective and less costly relative to ES ± mtDNA. The black curves above the x-axis suggest that the cost-effectiveness of GS relative to ES ± mtDNA improves as the difference in Dx yield increases.
In Approach 2, the genomic sequencing approach resulted in a mean-per-child cost of $9387 and in 38 definitive diagnoses in 100 children being tested. The mean-per-child cost in the conventional diagnostic pathway was estimated at $18,210, with 24 out of 100 children being definitively diagnosed. Thus, genomic sequencing resulted in a cost-saving of $8823 per child tested and in an additional 14 definitive diagnoses in 100 children. Genomic sequencing was again the dominant diagnostic strategy, with 100% probability of being cost-effective relative to the conventional diagnostic pathway (Figure S2). Based on the differences in the outcomes of genomic sequencing and conventional diagnostic care across the eight attributes included in our DCE (Table S5) [15], as identified in our analyses, the mean monetary value of the incremental benefits of genomic sequencing relative to conventional care was estimated at $5890 (95% CI: $5730−$6046). Genomic sequencing, therefore, results in a positive net benefit of $7887 (Approach 1a) and $14,713 (Approach 2).
Discussion
This study evaluated the cost-effectiveness of genomic sequencing in children with suspected MDs. Genomic sequencing was found to be less costly and more effective compared with conventional care in Australia, saving $1997 (Approach 1) to $8823 (Approach 2) per child tested, while leading to an additional 11 (Approach 1) to 14 (Approach 2) definitive diagnoses per 100 children tested relative to conventional care. Assuming an incidence rate of 1 per 10,000 live births for childhood-onset MDs [1], a diagnostic yield of 38%, and approximately 305,000 live births annually in Australia [24], this would suggest that approximately 80 children can be tested annually across Australia for MDs using genomic sequencing. Therefore, an implementation of genomic sequencing could translate to an annual cost-saving of $0.7 million and to an annual net societal benefit of $1.2 million, according to the outcomes of Approach 2, if genomic sequencing was implemented for the diagnosis of MDs.
To the best of our knowledge, this was the first economic evaluation assessing the cost-effectiveness of genomic sequencing in children with suspected MDs. Although economic evaluations of genomic sequencing have so far been limited, and with varying levels of methodological quality [12], evidence that genomic sequencing offers cost-savings relative to standard diagnostic care across different settings is increasingly emerging. In Australia, Yeung et al. [25] explored the cost-effectiveness of early genomic sequencing in pediatric patients with complex monogenic conditions compared with a matched historical cohort. The authors concluded that genomic sequencing resulted in cost-saving of AU$3602 (95% CI: $2520− $4685) and an additional 19 diagnoses out of 100 children tested. In Canada, Li et al. [26] evaluated the cost-effectiveness of genomic sequencing for unexplained developmental disabilities and multiple congenital anomalies. The authors concluded that genomic sequencing resulted in a cost-saving of CAN$2426 and an additional 23 diagnoses out of 100 patients tested.
The relevance of modeling over experimental and observational study designs has been well documented [22, 27]. The complexity of genomic sequencing coupled with ethical hurdles in conducting randomized controlled trials in the genomics space, and the rarity and heterogeneity of genetic conditions, essentially necessitates the use of decision modeling to inform reimbursement decisions. Decision modeling enables the synthesis of evidence from multiple sources, exploration of decision uncertainty and its effect on policy recommendation, comparisons of multiple alternatives, enabling optimal implementation, and inclusion of long-term and cascade, cost and outcome implications of testing, and thus, it is recommended for addressing the key challenges of genomic medicine [28, 29].
Our study benefited from primary clinical and economic data of an Australian-wide prospective cohort of children with suspected MDs, as well as a retrospective cohort of children that enabled incorporation of real-world data on the diagnostic investigations involved in conventional, nongenomic, pathways. The review of children’s medical files enabled identification of resource utilization with a high degree of accuracy, overcoming a key challenge in the economic evaluation of genomic sequencing in rare genetic diseases, which is modeling the costs and outcomes of standard care. Our study also benefited from a large DCE designed to elicit population preferences and values in Australia related to genomic sequencing [15]. This allowed us to incorporate the health and non-health value components of genomic sequencing [30], which has been one of the main challenges in the evaluation of genomics and precision medicine [29]. The complementary analyses, alongside the deterministic and probabilistic sensitivity analyses conducted, place confidence that the findings of this study can be generalizable to settings beyond Australia. Finally, another benefit of our study is the incorporation of costs associated with the clinical utility of genomic sequencing, an area where evidence is particularly lacking [31].
There are limitations, however, worth highlighting. Because the clinical phenotypes associated with MDs can be very broad, with considerable overlap with nonmitochondrial mendelian disorders, using clinical criteria scoring systems, including the Nijmegen criteria, for case selection, can at best be considered as a screening tool. Predictably, relying on just clinical criteria will result in lower genomic testing diagnostic yields. As protein biomarkers, such as GDF15 and FGF21 [32], become more readily available as additional screening tools, we can expect the diagnostic yield to improve significantly. Also, national funding agencies commonly base reimbursement decisions on health economics evidence generated using cost utility analyses, whereby quality-adjusted life-years (QALYs) represent the standard unit of outcome [33,34,35]. However, the relevance of QALYs in rare diseases has been questioned [36], and QALYs are not commonly used for the evaluation of genomic technologies [12], partly because of the large non-health benefits that are likely to be generated [30], and the pediatric population of interest [37]. Alternative methods for valuing the outcomes of genomic sequencing, such as DCEs, have been recommended [38], and are increasingly utilized [30]. Our analysis relied upon a large DCE conducted to elicit preferences for genomic sequencing from a large representative sample of the Australian general public. Our analyses incorporated evidence for healthcare resource utilization following on from genomic sequencing. However, estimates relied on a relatively small number of participants. Thus, it is likely that the cost estimates used for this parameter in the two approaches, could be different in another larger sample of participants. Nevertheless, deterministic and probabilistic sensitivity analyses performed for these parameters did not alter the conclusions drawn from the base-case analyses. Our analyses also modeled children with a clinical indication of MD and a Nijmegen score greater than 4. It may be likely that genomic sequencing is cost-effective for scores of 4 or below. Finally, there are significant diagnostic and economic opportunities involved in functional genomic studies conducted following a nondefinitive genomic diagnosis. Although some of these benefits were explored in our scenario analysis, studies that systematically evaluate the costs and outcomes of functional genomics are needed.
In conclusion, our study evaluated the cost-effectiveness and cost benefit of genomic sequencing relative to conventional diagnostic care for children with MDs from the perspective of the Australian healthcare system. The analyses performed concluded that genomic sequencing is cost-saving relative to traditional investigative approaches, while enabling more diagnoses to be made in a timely manner, offering substantial personal benefits to patients and their families. Implementation of genomic sequencing was found to lead to up to $0.7 million of savings per year to the Federal Government budget, freeing up resources that could be used to further improve population health. Our findings support the prioritization of genomic sequencing for children with mitochondrial disorders.
References
Skladal D, Halliday J, Thorburn DR. Minimum birth prevalence of mitochondrial respiratory chain disorders in children. Brain. 2003;126:1905–12.
Tan J, Wagner M, Stenton SL, Strom TM, Wortmann SB, Prokisch H, et al. Lifetime risk of autosomal recessive mitochondrial disorders calculated from genetic databases. EBioMedicine. 2020;54:102730.
Gorman GS, Chinnery PF, DiMauro S, Hirano M, Koga Y, McFarland R, et al. Mitochondrial diseases. Nat. Rev. Dis. Primers. 2016;2:16080.
Grier J, Hirano M, Karaa A, Shepard E, Thompson JLP. Diagnostic odyssey of patients with mitochondrial disease: results of a survey. Neurol. Genet. 2018;4:e230.
Parikh S, Goldstein A, Koenig MK, Scaglia F, Enns GM, Saneto R, et al. Diagnosis and management of mitochondrial disease: a consensus statement from the mitochondrial medicine society. Genet. Med. 2015;17:689–701.
Wortmann SB, Mayr JA, Nuoffer JM, Prokisch H, Sperl W. A guideline for the diagnosis of pediatric mitochondrial disease: the value of muscle and skin biopsies in the genetics era. Neuropediatrics. 2017;48:309–14.
Wu Y, Al-Janabi H, Mallett A, Quinlan C, Scheffer IE, Howell KB, et al. Parental health spillover effects of paediatric rare genetic conditions. Qual. Life Res. 2020;29:2445–54.
Payne K, Gavan SP, Wright SJ, Thompson AJ. Cost-effectiveness analyses of genetic and genomic diagnostic tests. Nat. Rev. Genet. 2018;19:235–46.
Best S, Stark Z, Phillips P, Wu Y, Long JC, Taylor N, et al. Clinical genomic testing: what matters to key stakeholders? Eur. J. Hum. Genet. 2020;28:866–73.
Clark MM, Stark Z, Farnaes L, Tan TY, White SM, Dimmock D, et al. Meta-analysis of the diagnostic and clinical utility of genome and exome sequencing and chromosomal microarray in children with suspected genetic diseases. NPJ Genom. Med. 2018;3:16.
Kohler JN, Turbitt E, Biesecker BB. Personal utility in genomic testing: a systematic literature review. Eur. J. Hum. Genet. 2017;25:662–8.
Schwarze K, Buchanan J, Taylor JC, Wordsworth S. Are whole-exome and whole-genome sequencing approaches cost-effective? A systematic review of the literature. Genet. Med. 2018;20:1122–30.
Stenton SL, Prokisch H. Genetics of mitochondrial diseases: Identifying mutations to help diagnosis. EBioMedicine. 2020;56:102784.
Goranitis I, Best S, Christodoulou J, Stark Z, Boughtwood T. The personal utility and uptake of genomic sequencing in pediatric and adult conditions: eliciting societal preferences with three discrete choice experiments. Genet. Med. 2020;22:1311–9.
Goranitis I, Best S, Stark Z, Boughtwood T, Christodoulou J. The value of genomic sequencing in complex pediatric neurological disorders: a discrete choice experiment. Genet. Med. 2021;23:155–62.
Morava E, van den Heuvel L, Hol F, de Vries MC, Hogeveen M, Rodenburg RJ, et al. Mitochondrial disease criteria: diagnostic applications in children. Neurology. 2006;67:1823–6.
Eddy DM, Hollingworth W, Caro JJ, Tsevat J, McDonald KM, Wong JB. Model transparency and validation: a report of the ISPOR-SMDM Modeling Good Research Practices Task Force–7. Med. Decis. Mak. 2012;32:733–43.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015;17:405–24.
Medicare Australia. Medicare Benefits Schedule (MBS) Item Statistics Reports. 2008. http://www.mbsonline.gov.au/internet/mbsonline/publishing.nsf/Content/FBE6CC5B217AC8DACA25859E0016F5A3/$File/July20_Complete%20MBS.pdf.
Pharmaceutical Benefits Advisory Committee (PBAC). Guidelines for preparing a submission to the Pharmaceutical Benefits Advisory Committee. Canberra, Australia: Australian Government, Department of Health and Ageing; 2008.
Small KA, Rosen HS. Applied welfare economics with discrete choice models. Econometrica: J. Econom. Soc. 1981;49:105–30.
Briggs A, Sculpher M, Claxton K. Decision modelling for health economic evaluation. Oxford: Oxford University Press; 2006.
Fenwick E, Claxton K, Sculpher M. Representing uncertainty: the role of cost-effectiveness acceptability curves. Health Econ. 2001;10:779–87.
Australian Bureau of Statistics (ABS). National, state and territory population statistics. ABS; 2020. https://www.abs.gov.au/statistics/people/population/national-state-and-territory-population/mar-2020.
Yeung A, Tan NB, Tan TY, Stark Z, Brown N, Hunter MF. et al. A cost-effectiveness analysis of genomic sequencing in a prospective versus historical cohort of complex pediatric patients. Genet. Med.2020;22:1986–93.
Li C, Vandersluis S, Holubowich C, Ungar WJ, Goh ES, Boycott KM. et al. Cost-effectiveness of genome-wide sequencing for unexplained developmental disabilities and multiple congenital anomalies. Genet. Med.2021;23:451–60.
Brennan A, Akehurst R. Modelling in health economic evaluation. Pharmacoeconomics. 2000;17:445–59.
Marshall DA, Grazziotin LR, Regier DA, Wordsworth S, Buchanan J, Phillips K, et al. Addressing challenges of economic evaluation in precision medicine using dynamic simulation modeling. Value Health. 2020;23:566–73.
Phillips KA, Deverka PA, Marshall DA, Wordsworth S, Regier DA, Christensen KD, et al. Methodological issues in assessing the economic value of next-generation sequencing tests: many challenges and not enough solutions. Value Health. 2018;21:1033–42.
Regier DA, Weymann D, Buchanan J, Marshall DA, Wordsworth S. Valuation of health and nonhealth outcomes from next-generation sequencing: approaches, challenges, and solutions. Value Health. 2018;21:1043–7.
Belsey J, Chaihorsky L, Currie G, Goranitis I, Marshall D. Global data access for solving rare disease: a health economics value framework. World Economic Forum. 2020. http://www3.weforum.org/docs/WEF_Global_Data_Access_for_Solving_Rare_Disease_Report_2020.pdf.
Lehtonen JM, Auranen M, Darin N, Sofou K, Bindoff L, Hikmat O. et al. Diagnostic value of serum biomarkers FGF21 and GDF15 compared to muscle sample in mitochondrial disease. J. Inherit. Metab. Dis.2020;44:469–80.
Canadian Agency for Drugs Technologies in Health (CADTH). Guidelines for the economic evaluation of health technologies. Ottawa, Canada; CADTH; 2006.
National Institute for Health and Clinical Excellence (NICE). Guide to the methods of technology appraisal. London, UK; NICE; 2013.
Pharmaceutical Benefits Advisory Committee (PBAC). Guidelines for preparing submissions to the Pharmaceutical Benefits Advisory Committee Canberra, Australia; PBAC; 2016.
Buchanan J, Wordsworth S. Evaluating the outcomes associated with genomic sequencing: a roadmap for future research. PharmacoEconomics Open. 2019;3:129–32.
De Civita M, Regier D, Alamgir AH, Anis AH, FitzGerald MJ, Marra CA. Evaluating health-related quality-of-life studies in paediatric populations. Pharmacoeconomics. 2005;23:659–85.
Grosse SD, Wordsworth S, Payne K. Economic methods for valuing the outcomes of genetic testing: beyond cost-effectiveness analysis. Genet. Med.2008;10:648–54.
Acknowledgements
Australian Genomics Health Alliance is funded by a National Health and Medical Research Council (NHMRC) grant (GNT: 1113531) and the Australian Government’s Medical Research Future Fund (MRFF), and a NHMRC research fellowship (GNT: 1102896). Philanthropic support from the Crane and Perkins families also funded this project. The research conducted at the Murdoch Children’s Research Institute was supported by the Victorian Government’s Operational Infrastructure Support Program. This work represents independent research and the views expressed are those of the authors and not necessarily those of the NHMRC or MRFF.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Wu, Y., Balasubramaniam, S., Rius, R. et al. Genomic sequencing for the diagnosis of childhood mitochondrial disorders: a health economic evaluation. Eur J Hum Genet 30, 577–586 (2022). https://doi.org/10.1038/s41431-021-00916-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-021-00916-8
This article is cited by
-
The value of genomic testing in severe childhood speech disorders
European Journal of Human Genetics (2024)
-
Determining the utility of diagnostic genomics: a conceptual framework
Human Genomics (2023)
-
QALYs and rare diseases: exploring the responsiveness of SF-6D, EQ-5D-5L and AQoL-8D following genomic testing for childhood and adult-onset rare genetic conditions in Australia
Health and Quality of Life Outcomes (2023)
-
2022: the year that was in the European Journal of Human Genetics
European Journal of Human Genetics (2023)
-
No gene to predict the future?
European Journal of Human Genetics (2022)
