
Abstract
Reanalysis of inconclusive exome/genome sequencing data increases the diagnosis yield of patients with rare diseases. However, the cost and efforts required for reanalysis prevent its routine implementation in research and clinical environments. The Solve-RD project aims to reveal the molecular causes underlying undiagnosed rare diseases. One of the goals is to implement innovative approaches to reanalyse the exomes and genomes from thousands of well-studied undiagnosed cases. The raw genomic data is submitted to Solve-RD through the RD-Connect Genome-Phenome Analysis Platform (GPAP) together with standardised phenotypic and pedigree data. We have developed a programmatic workflow to reanalyse genome-phenome data. It uses the RD-Connect GPAP’s Application Programming Interface (API) and relies on the big-data technologies upon which the system is built. We have applied the workflow to prioritise rare known pathogenic variants from 4411 undiagnosed cases. The queries returned an average of 1.45 variants per case, which first were evaluated in bulk by a panel of disease experts and afterwards specifically by the submitter of each case. A total of 120 index cases (21.2% of prioritised cases, 2.7% of all exome/genome-negative samples) have already been solved, with others being under investigation. The implementation of solutions as the one described here provide the technical framework to enable periodic case-level data re-evaluation in clinical settings, as recommended by the American College of Medical Genetics.
Similar content being viewed by others
Introduction
According to some estimations, around 350 million people worldwide may suffer from one of at least 7000 existing rare diseases (RDs) [1]. As 80% of RDs are thought to have a genetic origin [2, 3], the identification and characterisation of the molecular basis underlying these disorders is crucial for the establishment of a specific diagnosis and the subsequent identification of an optimal therapeutic approach.
The next generation sequencing (NGS) era has enabled cost-effective sequencing of RD patients’ exome or genome, bringing these approaches into diagnostics [4]. However, the identification and interpretation of disease-causing variants remains challenging. Indeed, the reported diagnostic yield for exome sequencing of RD patients with suspected monogenic disorders is around 20–60% depending on the type of disorder [5,6,7]. Undiagnosed cases can be re-approached by generating new genetic data using other techniques with more sensitivity than NGS for certain types of variants (e.g. arrays for large deletions or duplications) or re-sequencing the samples using other library strategies and sequencing protocols (e.g. whole genome sequencing, deep exon sequencing, a different exon capture kit, etc.).
Nevertheless, a negative result from NGS does not mean that the disease aetiology lies outside of the data already produced. In some cases, the variant is missed due to the bioinformatics analysis or incomplete phenotypic or family information. In other cases, the variant is not pinpointed because, at the time, the impact cannot be adequately assessed and/or the gene has not been yet associated with a certain function. However, technical developments and scientific understanding are constantly expanding, with new gene-disease associations increasing at an average rate of 250 per year (based on OMIM) and 9200 variant–disease associations being curated each year (based on HGMD) [8]. As a result, periodic data reanalysis and/or re-evaluation increases the diagnostic yield up to 10–12% [9,10,11], and the American College of Medical Genomics (ACMG) recommends variant-level re-evaluation and case-level reanalysis every 2 years [12].
While the scientific community extensively agrees on the benefits of periodic data reanalysis for RD patients, frequent re-evaluation of exomes/genomes is challenging in practice. The time-consuming effort required to identify the clinical record and re-assess segregated and unstructured genome-phenome data, together with the non-scalability of current solutions to reanalyse exponentially-growing datasets over time, preclude its implementation in research and clinical practice. Indeed, most clinical centres still do not include any re-evaluation approach in their routinely workflow as the benefit of identifying a new diagnosis is hardly unbalanced compared with the cost and efforts required for reanalysis. Therefore, innovative bioinformatics solutions are crucial to overcome some of these issues and facilitate iterative re-evaluation processes [11].
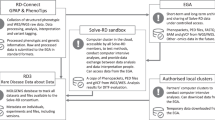
Solve-RD (http://solve-rd.eu/) aims to reveal the molecular cause underlying undiagnosed RDs [13]. One of the main goals of the project is to comprehensively reanalyse more than 19,000 phenotypically well characterised exome/genome negative datasets from unsolved patients with RDs submitted by European Reference Networks (ERNs). Besides the genomic data, the datasets include the phenotypic and pedigree information according to the RD-REAL (Rare Disease - REAnalysis Logistics) minimum information recommended for reanalysis [13]. All the existing RD-REAL datasets and the new ones generated by the project are being submitted to the RD-Connect Genome-Phenome Analysis Platform (GPAP, https://platform.rd-connect.eu/) as an entry point to the Solve-RD project.
The RD-Connect GPAP is an online platform that facilitates genome-phenome data analysis for RD diagnosis and gene discovery. Since datasets are submitted by many clinical researchers and are generated in different clinical centres and genomic facilities, the data are quite diverse at the source. To harmonise the information across all patients and relatives, the GPAP enables submission of pseudonymised phenotypic and clinical data using ontologies and standards such as the Human Phenotype Ontology (HPO) [14], the Orphanet Rare Disease Ontology (ORDO) [1], and the Online Mendelian Inheritance in Man database (OMIM) [2]. All the genomic data is processed through the same standardised pipeline [15] before being annotated and stored in an Elasticsearch database, which provides low-latency queries to enable fast access and ensure scalability.
Herein we describe a novel method that enables an automated, flexible, fast and iterative re-evaluation of thousands of genomic datasets using a programmatic access to the RD-Connect GPAP and we illustrate the utility of this procedure by reanalysing 4411 exome/genome negative index cases from the Solve-RD project. This approach has enabled the diagnosis of the first 120 cases within Solve-RD.
Patient and methods
Subjects
This study includes phenotypic and genomic data from 4703 affected individuals (4411 families) and 3690 unaffected relatives submitted to the RD-Connect GPAP as part of the Solve-RD project (http://solve-rd.eu/) [13] by four European Reference networks (the European Reference Networks for Rare Neurological Diseases (ERN-RND), Neuromuscular Diseases (ERN Euro NMD), Intellectual Disability and Congenital Malformations (ERN ITHACA) and Genetic Tumor Risk Syndromes (ERN GENTURIS), https://ec.europa.eu/health/ern_en), as well as two Undiagnosed Disease Programs (UDP Italy and UDP Spain). Clinical information was collated in a standard format using the HPO [14] for symptoms and the ORDO [1] for Clinical disorders. Each patient entry was associated with its corresponding submitting group and linked to its corresponding ERN or UDP. The responsibility of checking the data is suitable for submission to the RD-Connect GPAP and Solve-RD lies within the data submitter as required by their Code of Conduct and Data Sharing Policy, respectively. In some cases, individuals had to be re-consented prior to data submission. This study adheres to the principles set out in the Declaration of Helsinki.
Genomic data processing
4551 exome and 201 genome sequencing data (FastQ or BAM) derived from the 4703 affected individuals included in the Solve-RD freeze 1 dataset, were processed using the RD-Connect GPAP standardised analysis pipeline based upon GATK3.6 best practices and using the GRCh37 human reference, as described in ref. [15]. The resulting variants, including single nucleotide variants (SNVs), short insertions and deletions (InDels) and mtDNA variants (when captured) were annotated using VEP [16]. In addition, GnomAD [17], and ClinVar [18] were annotated with the latest versions available as for January 2020. Each dataset was associated with its corresponding phenotypic data and tagged with the name of the submitting ERN or UDP. Data are available to authorised users for analysis through the RD-Connect GPAP user interface (https://platform.rd-connect.eu/).
Programmatic access to genome-phenome datasets
Annotated genomic data is indexed in a non-relational ElasticSearch database engine (https://github.com/elastic/elasticsearch, GitHub - elastic/elasticsearch) connected to a Hadoop environment (Apache Software Foundation, https://hadoop.apache.org). Phenotypic data is stored in a local phenotypic database. Both genomic and phenotypic data are made computationally accessible through Application Programming Interface (API) endpoints, allowing automated queries through an in-house python package. To ensure secure and GDPR (General Data Protection Regulation) compliant data access for authorised users, the python package integrates a keycloak user authentication and permission management (github.com/keycloak/keycloak, GitHub - keycloak).
The GPAP’s API enables programmatic and flexible data analysis by (i) applying any type of filtering parameters according to the GPAP variants annotation (e.g. population frequencies, protein impact and in silico predictors), (ii) integrating standardised phenotypic information from each index case to create unique on-the-fly gene list for each of the experiments, (iii) filtering by specific gene lists according to the type of disorder (curated by ERNs, remote access to PanelApp from Genomics England or genes from any local or public database of interest), (iv) restraining the query filtering by homozygous regions in consanguineous cases or by specific regions of interest (e.g. regulatory regions) and (v) include segregation analysis based on the suspected inheritance and data from patient relatives introduced in the system.
Variant filtering parameters
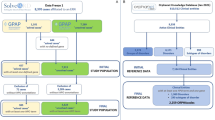
Variant filtering using the RD-Connect GPAP’s programmatic access described above was applied to identify candidate disease-causing SNVs and, InDels using the following parameters: [1] rare variants (observed population allele frequency <0.01 according to gnomAD and <0.02 according to the RD-Connect GPAP internal frequency), [2] specific gene list provided by the corresponding ERNs (euro-NMD, RND, ITHACA and GENTURIS) and [3] variant annotated as pathogenic or likely pathogenic for a specific disorder in ClinVar (v.13-01-2020). Apart from standard annotations (VEP), the resulting output file (one per ERN) was annotated with pseudonymised IDs, patient standardised phenotypic information (by extracting the corresponding HPOs and ORDO information entered in the system), candidate gene-disease associations (according to OMIM) [2], consanguinity reported and experimentally inferred (according to ref. [19]), gene constrain scores (pLI and o/e according to gnomAD v.2.1.1), ACMG computationally predicted clinical significance and criterias (using InterVar) [20] and when relevant, specific disease pathogenicity databases such as the VKGL database (https://www.vkgl.nl/nl/diagnostiek/vkgl-datashare-database) and the gene4denovo database [21]. The overall approach was designed by the Solve-RD SNV-indel working group from the Data Analysis Task Force (DATF) in collaboration with the corresponding disease expert groups [13] (Fig. 1).

Unsolved cases (RD-REAL datasets = phenotypic and genomic data) are submitted by Solve-RD members from the 4 core ERNs and the 2 UDPs participating in the project. Genomic data is processed through a standard analysis pipeline [15] and integrated with the phenotypic information in the RD-Connect GPAP. Analysis of the data using the programmatic approach described in this study is performed by the SNV-indel working group. The SNV-indel working group is one of the seven working groups established by the Solve-RD Data Analysis Task Force (DATF) to massively reanalyse data with different analytical approaches (e.g. CNV, somatic, meta-analysis, etc.) (http://solve-rd.eu/the-group/data-analysis-task-force/). The DATF involves data scientists and genomics experts from the project. Resulting candidate variants are submitted to the Data Interpretation Task Force (DITF), involving expert clinicians and geneticists for prioritisation and final interpretation. One DITF has been established for each of the core ERNs participating in the project (http://solve-rd.eu/the-group/data-interpretation-task-force-ditf/). DITF include or are in contact with case submitters to enable a final decision for a new patient diagnosis. Diagnosed cases are automatically updated in the system and the remaining unsolved cases are susceptible to re-enter a new round of analysis.
Variant prioritisation and data interpretation
Candidate variants from each case passing the filtering criteria are included in a single table to facilitate distribution across the Solve-RD network for evaluation and provision of feedback. The table is in MS Excel and has the same, or very similar, structure as the one provided by other Solve-RD DATF Working Groups for other type of genomic analyses. Solve-RD has organised ERNs clinical expertise in four dedicated Data Interpretation Task Forces (DITFs), one for each of the core ERNs. Results from the programmatic reanalysis performed were sent to the corresponding DITF members, a group of dedicated disease experts from the project who prioritised variants for further clinical assessment by data submitters (Fig. 1). Variant interpretation was then carried out in accordance with the criteria set by the ACMG guidelines [22] and the posterior ClinGen Sequence Variant Interpretation recommendations (https://www.clinicalgenome.org/working-groups/sequence-variant-interpretation/). The final feedback of variant pathogenicity for a specific clinical condition was determined by integrating patient assessment, variant evaluation and segregation, suspected inheritance, and clinical fit. Concerning family data available for segregation analyses, 28% of cases were submitted as trios (80% of them from ITHACA families), 68% were submitted as singletons (62% of them from RND) and 4% were from other family structures (Table 1).
Results
Programmatic reanalysis workflow
To enable automated and reproducible analysis and reanalysis of the Solve-RD data, we have developed a python package to execute queries through the RD-Connect API in a secure manner (Fig. 1). The parameters must be indicated in a configuration file, allowing a flexible (re)analysis environment covering very high to very low filtering stringencies and integrating patient clinical information through the use of computer readable standards (HPOs, ORDO, and OMIM) (Fig. 1). Options available for filtering include all annotations and features integrated in the RD-Connect GPAP from standard annotations (e.g. internal and external population allele frequencies) to more advanced features integrating clinical information to create patient specific on-the-fly gene lists (e.g. gene lists based on the HPOs entered for the index case). At the time being, the approach can detect SNVs and small InDels, including canonical splicing mutations. Other type of variants such as copy number variants will be integrated in the GPAP for filtering in future releases. In the meantime, Solve-RD has a specific DATF Working Group performing CNV analyses. Whenever relevant, the CNV variants are combined with the SNV/InDel results outside of the GPAP.
The queries are executed sequentially on the selected cases, enabling a scalable and tailored approach. The GPAP currently contains variants from 12,335 exomes and 638 genomes, distributed across 30 ElasticSearch instances in 12 server nodes (each with 2 octa-cores at 2.60 GHz, 256GB RAM and SSD disks). On these settings, each query requires 30 s per experiment on average.
The resulting variants are distributed to the respective DITF for variant prioritisation and interpretation (Fig. 1). After evaluation, the causative variants are tagged in the RD-Connect GPAP through the API or the graphical user-friendly interface. Unsolved cases may enter a new round of interpretation with a different combination of parameters and filters. New rounds of analysis are designed in collaboration with each of the DITF. Current approaches concern, for example, the identification of homozygous variants in homozygous stretches greater than 1 Mb for consanguineous cases or the identification of variants in known regulatory regions for specific patient cohorts (e.g. congenital myasthenic syndrome). Furthermore, other types of analyses are being done within Solve-RD, as indicated in ref. [13].
Application of the programmatic workflow for the reanalysis of undiagnosed rare disease patients
Bioinformatics reanalysis and the programmatic evaluation workflow were applied to all affected cases in the Solve-RD freeze 1 dataset [13]. In total, 4411 undiagnosed cases with heterogeneous genetic disorders were included: 1472 index cases referred as Intellectual disability (ERN-ITHACA), 2048 as Rare Neurological Disorder (ERN-RND), 616 as Neuromuscular Disorders (ERN-euroNMD), and 275 as Tumor Risk Syndromes (ERN-GENTURIS). Among the whole dataset, 55.7% of the cases were males and 44.3% females.
To minimise the interpretation burden for the DITFs, the first round of analysis was designed with very stringent parameters to allow the identification of clear candidates (”low-hanging fruit”) with known disease causality (Table 1, Fig. 1). All candidate variants were reported as “pathogenic” or “likely pathogenic” in ClinVar. Pathogenic variants are defined (based on the ACMG) as variants that directly contribute to the development of a disorder in a specific dosage sensitivity. The latter meaning that some pathogenic variants may not be fully penetrant or in the case of recessive or X-linked conditions, a single pathogenic variant may not be sufficient to cause disease on its own.
Total computational time for this analysis (including filtering and additional annotation steps for all 4411 experiments) was of 36 h and 45 min. The analysis yielded a total of 2593 candidates variants in 1785 index cases (40.4% of total cases, mean of 1.45 per individual) (Fig. 2A), which were distributed to the DITF. After each DITF applied additional prioritisation filters, a total of 678 variants from 566 index cases (31.7% of cases with identified variants; mean of 1.2 variants per individual) were sent to the referring clinical groups for final interpretation (Fig. 2A, Supplementary Table 1). Final interpretation was determined by integrating variant evaluation and patient phenotypic fit. The approach enabled to identify 124 causative variants leading to the diagnosis of 120 RD patients (21.2% of prioritised cases). Among the 124 causative variants identified (Supplementary Table 1), 68 (54.8%) were associated with an autosomal dominant disorder, 44 (35.6%) with an autosomal recessive disorder, 10 (8%) were X-linked, one (0.8%) in mitochondrial DNA and one (0.8%) was a mosaicism. In addition to the 120 diagnosed cases, 26 variants from 25 index cases are still under evaluation (segregation analysis, clinical re-evaluation, SANGER validation, etc.) by the clinical submitting groups (Fig. 2A, C). For an additional 87 index cases, 103 heterozygous variants in phenotype-related candidate genes associated with autosomal recessive disorders were identified. In some of those cases, additional analyses or new data might identify another variant that could finally diagnose the case.

A Filtration, prioritisation and interpretation workflow (numbers refer to index cases). B Number of variants per case submitted to DITFs for prioritisation and resulting number of variants submitted for interpretation. C Variants interpretation results from prioritised cases per type of disorder (numbers refer to variants). D Number of causative variants identified according to the year the corresponding gene (grey) or variant (yellow) was first described in the literature as disease-causing (according to OMIM) or pathogenic (according to ClinVar).
We hypothesised that several cases could have remained undiagnosed when they were originally analysed because knowledge on a specific gene function or variant impact might have been lacking at the time. To further investigate this point, we retrieved, for each of the causative variants, the date when the corresponding gene was first associated with a disease and a pathogenic variant for a specific clinical condition reported in ClinVar (Fig. 2D). In total, 16 (13%) newly identified causative variants were found in genes associated with disorders since 2017 (2 years since data was sent for reanalysis), 11 (9%) between 2015 and 2016, 39 (31%) between 2010 and 2014 and 60 (47%) before 2010. Concerning the clinical significance of the variant, 48 (39%) newly identified causative variants were submitted as pathogenic for a specific disorder to ClinVar since 2017 (2 years since data was sent for reanalysis), 27 (21%) between 2015 and 2016, 40 (31%) between 2010–2014 and 11 (9%) before 2010.
Among the 26 homozygous causative variants (Supplementary Table 2), 15 were identified in experimentally determined consanguineous probands according to ref. [19], being 13 of them within a homozygous stretch of more than 1 Mb (Supplementary Table 2). In order to discard possible false homozygous calls due to a hypothetic heterozygous deletion of the region covering the causative variant in non-consanguineous probands, we cross-checked CNV results provided by the Solve-RD DATF. No deletions in the region of interest were detected.
Discussion
Constant improvement of bioinformatics methods and advances in genomic understanding to identify and interpret variants highlight the need to periodically re-evaluate unsolved exome/genome cases as stressed by the ACMG [12]. However, to date, the benefit of identifying a new diagnosis in clinical environments is hardly unbalanced compared with the efforts required for re-evaluation. In this study, we present a rapid, scalable and cost-effective approach to programmatically (re)analyse thousands of structured genome-phenome RD-REAL datasets from undiagnosed cases collated as part of the Solve-RD project [13].
We have set up a programmatic system based on a python package to query structured genome-phenome data from the RD-Connect GPAP through its dedicated API. Only sample IDs and filtering parameters need to be defined in the system before attempting a new (re)analysis. Then, the fully automated approach enables to intelligently and flexibly filter genomic data based on clinical, familial, biological and genomic quality information in a rapid (30 s per experiment on average) and massive way (currently >4400 samples tested). The big-data technologies upon which the RD-Connect GPAP is built enable systems to grow by adding more resources as needed. The described approach will allow for the (re)analysis of all the 19,000 exome/genome datasets that Solve-RD aims to collect and the new data it is producing [13].
Despite the use of cutting-edge technologies, and that experts are able to re-evaluate hundreds of cases with the key information at sight, clinical interpretation remains a manual process. In order to facilitate and reduce interpretation efforts, the programmatic output is provided in a meaningful way, integrating relevant genomic, biological and clinical information for referring clinicians and clinical scientists to perform this final step. Results can be enriched with additional annotations and can also include the link to the specific query in the RD-Connect GPAP, enabling the users to explore the variant within a graphical user-interface. We tested the approach with the 4411 affected cases from the first Solve-RD data freeze. All those cases were well characterised and had an exome/genome that had been thoroughly analysed without success. Only the first “low-hanging fruit” filtering approach for rare known pathogenic variants (according to ClinVar) in known disease-causing genes already allowed us to solve 120 undiagnosed index cases (21.2% of prioritised cases). The approach included the use of dedicated ERN associated gene lists to focus on diseases under investigation and limiting the risks of secondary findings. Heterozygous potential candidate variants for autosomal recessive disorders were also identified in 15.3% of the prioritised cases.
The overall positive results obtained from the prioritised variants of this “low-hanging fruit” reanalysis approach can be attributed to several factors. The original exome/genome data reanalysed in this study were sequenced by different centers at different times. This means that the original analyses (including mapping, variant calling, annotation and filtering) were performed with a variety of different tools and databases, likely using different versions and parameters. In addition, the human genome reference used might have been different even if with small changes (e.g. with or without viral and/or decoy sequences). Therefore, the pipeline used in Solve-RD will be in almost all cases somewhat different than the one used in the original analysis, which might have had an effect in unveiling previously undetected variants (e.g ref. [23]). Furthermore, scientific knowledge improves with time, enabling to identify previously undetected associations. In our study, 13% of the newly identified causative variants were in genes not associated with disease in the 2 years prior to reanalysis (described since 2017) and 39% were variants not reported as (likely) pathogenic for similar clinical manifestations at that time. If we assume reanalysis was not performed in the previous 4 years prior to submission, these values increase up to 22% for new disease-causing genes and 60% for newly reported pathogenic variants (e.g. ref. [24] and ref. [25]). Finally, standardised clinical information using HPO, ORDO and OMIM combined with different filtering approaches helped prioritise causative variants in atypical phenotypes (e.g. ref. [25]). This result is aligned with previous studies in the RD-Connect GPAP on a cohort of patients with rare neuromuscular disorders reporting the importance of deep and accurate phenotyping for variant prioritisation [26]. For cases remaining undiagnosed, it might be useful to keep updating the patient phenotypic descriptions with new observations, as this might help identify additional candidate pathogenic variants for the disease and increase specificity of the filtering step, thus lowering the time necessary for variant re-evaluation. In this sense, the RD-Connect GPAP facilitates updating the patient records through its phenotypic module. Remarkably, the interpretation of several causative variants identified in complex genes or regions was possible thanks to the multidisciplinary team of RD experts involved (e.g. ref. [24]).
This first “low-hanging fruit” automated approach managed to solve 2.7% of all clinically heterogeneous undiagnosed and previously negative-exome/genome cases in <37 h of computational time. The flexibility of the system described herein is now being applied to additional strategic reanalyses, varying parameters stringencies and contributing to increase the diagnostic yield. New approaches will focus on the identification of mtDNA variants using specific variant callers [27] or the inclusion of additional clinical resources such as HGMD [8] or Varsome [28]. Indeed, the GPAP already provides direct links to those clinical databases to facilitate variant interpretation and another re-evaluation approach relying on the HGMD database is planned for filtering by (likely) pathogenic variants based on the data available by the user’s license. Several other Solve-RD working groups, focused on the identification of other types of variants or analysis strategies (e.g. copy number variants, repeat expansions or de novo analyses) and/or integrating new –omics generated within the project (e.g. RNA-seq, long read WGS) are joining efforts to unravel additional molecular causes underlying RDs [13].
In comparison and similarly to other iterative reanalysis strategies [10, 29], our approach has three main advantages and time-saving points for clinicians and clinical scientists. First, experts do not need to re-annotate and filter manually with different strategies thousands of cases. Second, they only need to re-evaluate the cases for which at least one candidate variants has been proposed (40.4% of cases in our study). Third, the output file contains all the cases with candidate variants identified and includes key information for their preliminary evaluation.
This method could be adapted to any diagnostic (re)analysis workflow and extended to the whole RD-Connect dataset (currently >13,000 samples) or any subset of interest. Data can be periodically re-evaluated with no additional cost and according to any predefined period of time (e.g. every 6 months or once a year) or after relevant method improvements or database updates. This strategy reduces reanalysis costs and experts’ time-consuming efforts while offering a solution to three out of the four key elements to reinterpret genetic data recently raised by ref. [30]: data storage and re-access, initiation of routine reinterpretation and reinterpretation with novel information.
In summary, we have developed a scalable, cost-effective programmatic approach to drastically decrease turnaround time and effort for periodic data reanalysis. We have illustrated the usefulness of the system by revealing the molecular bases of 120 previously undiagnosed patients with RDs within Solve-RD. This methodology can be implemented systematically in a clinical diagnostic setting for periodic case-level data re-evaluation, as recommended by the ACMG [12].
Change history
16 August 2021
A Correction to this paper has been published: https://doi.org/10.1038/s41431-021-00934-6
References
Nguengang Wakap S, Lambert DM, Olry A, Rodwell C, Gueydan C, Lanneau V, et al. Estimating cumulative point prevalence of rare diseases: analysis of the Orphanet database. Eur J Hum Genet. 2020;28:165–73.
Amberger JS, Bocchini CA, Schiettecatte F, Scott AF, Hamosh A. OMIM.org: online Mendelian Inheritance in Man (OMIM®), an online catalog of human genes and genetic disorders. Nucleic Acids Res. 2015;43:D789–98.
Amberger JS, Hamosh A. Searching Online Mendelian Inheritance in Man (OMIM): a Knowledgebase of Human Genes and Genetic Phenotypes. Curr Protoc Bioinforma. 2017;58:1.2.1–1.2.12.
Boycott KM, Hartley T, Biesecker LG, Gibbs RA, Innes AM, Riess O, et al. A Diagnosis for All Rare Genetic Diseases: the Horizon and the Next Frontiers. Cell 2019;177:32–7.
Wright CF, Fitzgerald TW, Jones WD, Clayton S, McRae JF, van Kogelenberg M, et al. Genetic diagnosis of developmental disorders in the DDD study: a scalable analysis of genome-wide research data. Lancet 2015;385:1305–14.
Farwell KD, Shahmirzadi L, El-Khechen D, Powis Z, Chao EC, Tippin Davis B, et al. Enhanced utility of family-centered diagnostic exome sequencing with inheritance model-based analysis: results from 500 unselected families with undiagnosed genetic conditions. Genet Med. 2015;17:578–86.
Stark Z, Tan TY, Chong B, Brett GR, Yap P, Walsh M, et al. A prospective evaluation of whole-exome sequencing as a first-tier molecular test in infants with suspected monogenic disorders. Genet Med. 2016;18:1090–6.
Wenger AM, Guturu H, Bernstein JA, Bejerano G. Systematic reanalysis of clinical exome data yields additional diagnoses: implications for providers. Genet Med. 2017;19:209–14.
Salfati EL, Spencer EG, Topol SE, Muse ED, Rueda M, Lucas JR, et al. Re-analysis of whole-exome sequencing data uncovers novel diagnostic variants and improves molecular diagnostic yields for sudden death and idiopathic diseases. Genome Med. 2019;11:83.
Liu P, Meng L, Normand EA, Xia F, Song X, Ghazi A, et al. Reanalysis of Clinical Exome Sequencing Data. N Engl J Med. 2019;380:2478–80.
Baker SW, Murrell JR, Nesbitt AI, Pechter KB, Balciuniene J, Zhao X, et al. Automated Clinical Exome Reanalysis Reveals Novel Diagnoses. J Mol Diagn. 2019;21:38–48.
Deignan JL, Chung WK, Kearney HM, Monaghan KG, Rehder CW, Chao EC, ACMG Laboratory Quality Assurance Committee. Points to consider in the reevaluation and reanalysis of genomic test results: a statement of the American College of Medical Genetics and Genomics (ACMG). Genet Med. 2019;21:1267–70.
Zurek B, Ellwanger K, Vissers L, Schüle R, Synofzik M, Töpf A, et al. Solve-RD: systematic Pan-European data sharing and collaborative analysis to solve Rare Diseases. Accepted to EJHG - 698-20-EJHG.
Köhler S, Carmody L, Vasilevsky N, Jacobsen JOB, Danis D, Gourdine JP, et al. Expansion of the Human Phenotype Ontology (HPO) knowledge base and resources. Nucleic Acids Res. 2019;47:D1018–27.
Laurie S, Fernandez-Callejo M, Marco-Sola S, Trotta JR, Camps J, Chacón A. From Wet-Lab to Variations: concordance and Speed of Bioinformatics Pipelines for Whole Genome and Whole Exome Sequencing. Hum Mutat. 2016;37:1263–71.
McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GR, Thormann A, et al. The Ensembl Variant Effect Predictor. Genome Biol. 2016;17:122.
Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581:434–43.
Landrum MJ, Lee JM, Benson M, Brown G, Chao C, Chitipiralla S, et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2016;44:D862–8.
Matalonga L, Laurie S, Papakonstantinou A, Piscia D, Mereu E, Bullich G, et al. Improved Diagnosis of Rare Disease Patients through Systematic Detection of Runs of Homozygosity. J Mol Diagn. 2020;22:1205–15.
Li Q, Wang K. InterVar: clinical Interpretation of Genetic Variants by the 2015 ACMG-AMP Guidelines. Am J Hum Genet. 2017;100:267–80.
Zhao G, Li K, Li B, Wang Z, Fang Z, Wang X, et al. Gene4Denovo: an integrated database and analytic platform for de novo mutations in humans. Nucleic Acids Res. 2020;48:D913–26.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24.
te Paske, Garcia Pelaez J, Matalonga L, Starzynska T, Jakubowska A, Solve-RD-GENTURIS group et al. A Mosaic PIK3CA Variant in Young Adult with Diffuse Gastric Cancer: Case Report. Accepted to EJHG- 704-20-EJHG.
Töpf A, Pyle A, Griffin H, Matalonga L, Schon K, Solve-RD SNV-indel working group, et al. Exome reanalysis and proteomic profiling identified TRIP4 as a novel cause of pontocerebellar hypoplasia and spinal muscular atrophy (PCH1). Accepted to EJHG- 700-20-EJHG.
Schüle R, Timmann D, Erasmus C, Reichbauer J, Wayand M, van de Warrenburg B, et al. Solving unsolved rare neurological diseases – a Solve-RD viewpoint. Submitted to EJHG- 705-20-EJHG.
Thompson R, Papakonstantinou Ntalis A, Beltran S, Töpf A, de Paula Estephan E, et al. Increasing phenotypic annotation improves the diagnostic rate of exome sequencing in a rare neuromuscular disorder. Hum Mutat. 2019;40:1797–812.
de Boer E, Ockeloen CW, Matalonga L, Horvath R, Solve-RD SNV-indel working group, Rodenburg RJ, et al. A pathogenic MT-TL1 variant identified by whole exome sequencing in an individual with unexplained intellectual disability, epilepsy and spastic tetraparesis. Accepted to EJHG- 699-20-EJHG.
Kopanos C, Tsiolkas V, Kouris A, Chapple CE, Albarca Aguilera M, et al. VarSome: the human genomic variant search engine. Bioinformatics 2019;35:1978–80.
Wright CF, McRae JF, Clayton S, Gallone G, Aitken S, FitzGerald TW, et al. Making new genetic diagnoses with old data: iterative reanalysis and reporting from genome-wide data in 1,133 families with developmental disorders. Genet Med. 2018;20:1216–23.
Appelbaum PS, Parens E, Berger SM, Chung WK, Burke W. Is there a duty to reinterpret genetic data? The ethical dimensions. Genet Med. 2020;22:633–9.
Acknowledgements
The authors would like to thank the Solve-RD SNV-indel working group for its support on setting up the analysis and Solve-RD-DITF-GENTURIS, Solve-RD-DITF-ITHACA, Solve-RD-DITF-RND, Solve-RD-DITF-euroNMD, UDP-Spain and UDP-Italy for providing feedback on data interpretation.
Solve-RD SNV-indel working group:
Enzo Cohen13, Isabel Cuesta14, Daniel Danis15, Anne-Sophie Denommé-Pichon16,17,18, Yannis Duffourd16,18, Christian Gilissen5,7, Mridul Johari19, Steven Laurie1, Shuang Li20, Leslie Matalonga1, Isabelle Nelson13, Sophia Peters21, Ida Paramonov1, Sivakumar Prasanth22, Peter Robinson15, Karolis Sablauskas5,7, Marco Savarese19, Wouter Steyaert5,7, Ana Töpf4, Joeri K. van der Velde20, Antonio Vitobello16
Solve-RD DITF-GENTURIS:
Stefan Aretz21,23, Gabriel Capella5,7, Richarda M. de Voer5,7, Gareth Evans24, Jose Garcia Pelaez25,26, Elke Holinski-Feder27, Nicoline Hoogerbrugge5,7, Andreas Laner27, Carla Oliveira25,26,28, Andreas Rump29, Evelin Schröck29, Anna Katharina Sommer21, Verena Steinke-Lange27, Iris te Paske5,7, Marc Tischkowitz30, Laura Valle31
Solve-RD DITF-ITHACA:
Siddharth Banka32,33, Elisa Benetti34, Giorgio Casari35,36, Andrea Ciolfi37, Jill Clayton-Smith32,33, Bruno Dallapiccola37, Elke de Boer5,6, Anne-Sophie Denommé-Pichon16,17,38, Kornelia Ellwanger9,39, Laurence Faivre16,18,40, Christian Gilissen5,7, Holm Graessner9,39, Tobias B. Haack9, Anna Hammarsjö41, Marketa Havlovicova42, Alexander Hoischen5,8,33, Anne Hugon43, Adam Jackson40, Tjitske Kleefstra5,6, Anna Lindstrand41, Estrella López-Martín14, Milan Macek Jr42, Leslie Matalonga1, Manuela Morleo36, Vicenzo Nigro36, Ann Nordgren41, Maria Pettersson41, Michele Pinelli36, Simone Pizzi37, Manuel Posada14, Francesca Clementina Radio44, Alessandra Renieri34,45,46, Caroline Rooryck47, Lukas Ryba42, Martin Schwarz42, Marco Tartaglia37, Christel Thauvin16,40, Annalaura Torella35,36, Aurélien Trimouille38, Alain Verloes43,48, Lisenka Vissers5,6, Antonio Vitobello16, Pavel Votypka42, Klea Vyshka43,48, Birte Zurek9,39
Solve-RD DITF-euroNMD:
Ana Töpf4, Jonathan Baets49,50,51, Danique Beijer49,50, Gisèle Bonne13, Enzo Cohen13, Judith Cossins52, Teresinha Evangelista13, Alessandra Ferlini53, Peter Hackman54, Michael G. Hanna55, Rita Horvath56, Henry Houlden55, Mridul Johari54, Jarred Lau57, Hanns Lochmüller1,57,58,59,60, William L. Macken55, Francesco Musacchia35,36, Andres Nascimento61, Daniel Natera-de Benito61, Vincenzo Nigro35,36, Giulio Piluso35, Veronica Pini62, Robert D. S. Pitceathly55, Kiran Polavarapu57,60, Pedro M. Rodriguez Cruz52,63, Anna Sarkozy62, Marco Savarese54, Rita Selvatici53, Rachel Thompson57, Annalaura Torella35,36, Bjarne Udd54, Liedewei Van de Vondel50,51, Jana Vandrovcova55, Irina Zaharieva62
Solve-RD DITF-RND:
Jonathan Baets50,51,64, Peter Balicza65, Patrick Chinnery23, Alexandra Dürr66,67,68, Tobias Haack9, Holger Hengel2,3, Rita Horvath56, Henry Houlden55, Erik-Jan Kamsteeg5, Christoph Kamsteeg5, Katja Lohmann69, Alfons Macaya70, Anna Marcé-Grau70, Ales Maver39, Judit Molnar65, Alexander Münchau69, Borut Peterlin71, Olaf Riess9,39, Ludger Schöls2,3, Rebecca Schüle-Freyer2,3, Giovanni Stevanin66,67,68,72,73, Matthis Synofzik2,3, Vincent Timmerman74,75, Bart van de Warrenburg6, Nienke van Os6,76, Jana Vandrovcova55, Melanie Wayand2,3, Carlo Wilke2,3
The Solve-RD Consortia:
Olaf Riess9,39, Tobias B. Haack9, Holm Graessner9,39, Birte Zurek9,39, Kornelia Ellwanger9,39, Stephan Ossowski9, German Demidov9, Marc Sturm9, Julia M. Schulze-Hentrich9, Rebecca Schüle2,3, Christoph Kessler2,3, Melanie Wayand2,3, Matthis Synofzik2,3, Carlo Wilke2,3, Andreas Traschütz2,3, Ludger Schöls2,3, Holger Hengel2,3, Peter Heutink2,3, Han Brunner5,6,77, Hans Scheffer5,77, Nicoline Hoogerbrugge5,7, Alexander Hoischen5,7,8, Peter A. C. ’t Hoen7,78, Lisenka E. L. M. Vissers5,6, Christian Gilissen5,7, Wouter Steyaert5,7, Karolis Sablauskas5, Richarda M. de Voer5,7, Erik-Jan Kamsteeg5, Bart van de Warrenburg6,76, Nienke van Os6,76, Iris te Paske5,7, Erik Janssen5,7, Elke de Boer5,6, Marloes Steehouwer5, Burcu Yaldiz5, Tjitske Kleefstra5,6, Anthony J. Brookes79, Colin Veal79, Spencer Gibson79, Marc Wadsley79, Mehdi Mehtarizadeh79, Umar Riaz79, Greg Warren79, Farid Yavari Dizjikan79, Thomas Shorter79, Ana Töpf4, Volker Straub4, Chiara Marini Bettolo4, Sabine Specht4, Jill Clayton-Smith24, Siddharth Banka24,32, Elizabeth Alexander24, Adam Jackson24, Laurence Faivre16,17,18,80,81, Christel Thauvin16,18,80,81, Antonio Vitobello16, Anne-Sophie Denommé-Pichon16, Yannis Duffourd16,18, Emilie Tisserant16, Ange-Line Bruel16, Christine Peyron82,83, Aurore Pélissier83, Sergi Beltran1,11, Ivo Glynne Gut11, Steven Laurie11, Davide Piscia11, Leslie Matalonga11, Anastasios Papakonstantinou11, Gemma Bullich11, Alberto Corvo11, Carles Garcia11, Marcos Fernandez-Callejo11, Carles Hernández11, Daniel Picó11, Ida Paramonov11, Hanns Lochmüller11, Gulcin Gumus84, Virginie Bros-Facer85, Ana Rath86, Marc Hanauer86, Annie Olry86, David Lagorce86, Svitlana Havrylenko86, Katia Izem86, Fanny Rigour86, Giovanni Stevanin66,67,68,72,73, Alexandra Durr67,68,72,87, Claire-Sophie Davoine67,68,72,73, Léna Guillot-Noel67,68,72,73, Anna Heinzmann67,68,72,88, Giulia Coarelli67,68,72,88, Gisèle Bonne13, Teresinha Evangelista13, Valérie Allamand13, Isabelle Nelson13, Rabah Ben Yaou13,89,90, Corinne Metay13,91, Bruno Eymard13,89, Enzo Cohen13, Antonio Atalaia13, Tanya Stojkovic13,89, Milan Macek Jr.42, Marek Turnovec42, Dana Thomasová42, Radka Pourová Kremliková42, Vera Franková42, Markéta Havlovicová42, Vlastimil Kremlik42, Helen Parkinson92, Thomas Keane92, Dylan Spalding92, Alexander Senf92, Peter Robinson15, Daniel Danis15, Glenn Robert93, Alessia Costa93, Christine Patch93,94, Mike Hanna22, Henry Houlden95, Mary Reilly22, Jana Vandrovcova95, Francesco Muntoni62,96, Irina Zaharieva62, Anna Sarkozy62, Vincent Timmerman74,75, Jonathan Baets50,51,64, Liedewei Van de Vondel64,75, Danique Beijer64,75, Peter de Jonghe51,75, Vincenzo Nigro35,36, Sandro Banfi35,36, Annalaura Torella35, Francesco Musacchia35,36, Giulio Piluso35, Alessandra Ferlini53, Rita Selvatici53, Rachele Rossi53, Marcella Neri53, Stefan Aretz21,23, Isabel Spier21,23, Anna Katharina Sommer21, Sophia Peters21, Carla Oliveira25,26,28, Jose Garcia Pelaez25,26, Ana Rita Matos25,26, Celina São José25,26, Marta Ferreira25,26, Irene Gullo25,26,28, Susana Fernandes25,97, Luzia Garrido98, Pedro Ferreira25,26,99, Fátima Carneiro25,26,28, Morris A. Swertz20, Lennart Johansson20, Joeri K. van der Velde20, Gerben van der Vries20, Pieter B. Neerincx20, Dieuwke Roelofs-Prins20, Sebastian Köhler100, Alison Metcalfe93,101, Alain Verloes43,48, Séverine Drunat43,48, Caroline Rooryck47, Aurelien Trimouille38, Raffaele Castello36, Manuela Morleo36, Michele Pinelli36, Alessandra Varavallo36, Manuel Posada De la Paz14, Eva Bermejo Sánchez14, Estrella López Martín14, Beatriz Martínez Delgado14, F. Javier Alonso García de la Rosa14, Andrea Ciolfi37, Bruno Dallapiccola37, Simone Pizzi37, Francesca Clementina Radio37, Marco Tartaglia37, Alessandra Renieri34,45,46, Elisa Benetti34, Peter Balicza102, Maria Judit Molnar102, Ales Maver103, Borut Peterlin103, Alexander Münchau104, Katja Lohmann104, Rebecca Herzog104, Martje Pauly104, Alfons Macaya105, Anna Marcé-Grau105, Andres Nascimiento Osorio106, Daniel Natera de Benito106, Hanns Lochmüller60,107,108, Rachel Thompson60,108, Kiran Polavarapu60, David Beeson63, Judith Cossins63, Pedro M. Rodriguez Cruz63, Peter Hackman109, Mridul Johari109, Marco Savarese109, Bjarne Udd109,110,111, Rita Horvath112, Gabriel Capella113, Laura Valle113, Elke Holinski-Feder114, Andreas Laner114, Verena Steinke-Lange114, Evelin Schröck115, Andreas Rump115,116
Funding
The Solve-RD project has received funding from the European Union’s Horizon 2020 research and innovation programme under grant agreement No 779257. Data were analysed using the RD‐Connect Genome‐Phenome Analysis Platform, which received funding from EU projects RD‐Connect, Solve-RD and EJP-RD (grant numbers FP7 305444, H2020 779257, H2020 825575), Instituto de Salud Carlos III (grant numbers PT13/0001/0044, PT17/0009/0019; Instituto Nacional de Bioinformática, INB) and ELIXIR Implementation Studies. We acknowledge support of the Spanish Ministry of Economy, Industry and Competitiveness (MEIC) to the EMBL partnership, the Centro de Excelencia Severo Ochoa and the CERCA Programme/Generalitat de Catalunya. We also acknowledge the support of the Generalitat de Catalunya through Departament de Salut and Departament d’Empresa i Coneixement and the Co-financing by the Spanish Ministry of Economy, Industry and Competitiveness (MEIC) with funds from the European Regional Development Fund (ERDF) corresponding to the 2014-2020 Smart Growth Operating Program.
Author information
Authors and Affiliations
Consortia
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Members of the Solve-RD SNV-indel working group, Solve-RD DITF-GENTURIS, Solve-RD DITF-ITHACA, Solve-RD DITF-euroNMD, Solve-RD DITF-RND, and Solve-RD Consortia are listed below Acknowledgements.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Matalonga, L., Hernández-Ferrer, C., Piscia, D. et al. Solving patients with rare diseases through programmatic reanalysis of genome-phenome data. Eur J Hum Genet 29, 1337–1347 (2021). https://doi.org/10.1038/s41431-021-00852-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-021-00852-7
This article is cited by
-
Phenotypic similarity-based approach for variant prioritization for unsolved rare disease: a preliminary methodological report
European Journal of Human Genetics (2024)
-
Systematic reanalysis of genomic data by diagnostic laboratories: a scoping review of ethical, economic, legal and (psycho)social implications
European Journal of Human Genetics (2024)
-
Variants in mitochondrial disease genes are common causes of inherited peripheral neuropathies
Journal of Neurology (2024)
-
International Undiagnosed Diseases Programs (UDPs): components and outcomes
Orphanet Journal of Rare Diseases (2023)
-
Updated Stroke Gene Panels: Rapid evolution of knowledge on monogenic causes of stroke
European Journal of Human Genetics (2023)
