
Abstract
The Koolen-de Vries syndrome (KdVS) is a multisystem syndrome with variable facial features caused by a 17q21.31 microdeletion or KANSL1 truncating variant. As the facial gestalt of KdVS has resemblance with the gestalt of the 22q11.2 deletion syndrome (22q11.2DS), we assessed whether our previously described hybrid quantitative facial phenotyping algorithm could distinguish between these two syndromes, and whether there is a facial difference between the molecular KdVS subtypes. We applied our algorithm to 2D photographs of 97 patients with KdVS (78 microdeletions, 19 truncating variants (likely) causing KdVS) and 48 patients with 22q11.2DS as well as age, gender and ethnicity matched controls with intellectual disability (n = 145). The facial gestalts of KdVS and 22q11.2DS were both recognisable through significant clustering by the hybrid model, yet different from one another (p = 7.5 × 10−10 and p = 0.0052, respectively). Furthermore, the facial gestalts of KdVS caused by a 17q21.31 microdeletion and KANSL1 truncating variant (likely) causing KdVS were indistinguishable (p = 0.981 and p = 0.130). Further application to three patients with a variant of unknown significance in KANSL1 showed that these faces do not match KdVS. Our data highlight quantitative facial phenotyping not only as a powerful tool to distinguish syndromes with overlapping facial dysmorphisms but also to establish pathogenicity of variants of unknown clinical significance.
Similar content being viewed by others

Introduction
The Koolen-de Vries syndrome (KdVS) (OMIM #610443), also known as 17q21.31 microdeletion syndrome or KANSL1-related intellectual disability syndrome, is a multisystem disorder characterised by developmental delay, intellectual disability, distinctive facial features, hypotonia, epilepsy, amiable behavior and congenital malformations in multiple other organ systems [1,2,3,4,5,6].
The clinical spectrum and the facial appearance of KdVS are variable [4] and the syndrome can be caused either by a microdeletion in the chromosomal region 17q21.31 or by a truncating variant in the KAT 8 regulatory NSL complex unit 1 (KANSL1) gene (NG_032784.1) [5, 6]. A previous clinical study comparing the clinical phenotype of KdVS patients with either the deletion or the a truncating variant in KANSL1 (33 versus 12 patients respectively) showed a similarity in phenotypic presentation, including the facial phenotype, for both molecular subtypes. The facial phenotype, however, was based on the (subjective) assessment by the clinician, and prior knowledge of the molecular diagnosis [7].
The most frequently reported findings in the KdVS—developmental delay and childhood hypotonia—are common and relatively nonspecific indications for genetic testing. The differential diagnosis of patients with KdVS consists of several other syndromes such as 22q11.2 deletion syndrome (OMIM #611867), Prader-Willi syndrome (OMIM #176270) and Fragile X syndrome (OMIM #300624) [4, 3].
Especially the 22q11.2 deletion syndrome (22q11.2DS) (the most common cause of DiGeorge syndrome and velocardiofacial syndrome) can have similar facial dysmorphisms—such as a long face, malar flatness, hooded eyelids resulting in the appearance of narrow palpebral fissures, and hypoplastic alae nasi accentuating the bulbous nasal tip often accompanied by a nasal dimple/crease/hemangioma—as KdVS [8,9,10]. In clinical practice, when taking the phenotype of a specific patient into account, it is usually possible to distinguish between these two conditions.
Several studies using facial recognition tools based on 2D profile photos have meanwhile shown their successes to help clinicians in diagnosing intellectual disability syndromes [11,12,13]. More recently, we adjusted two existing models to a hybrid model for quantitative facial phenotyping [14]. This optimised tool combines the output of two computer vision algorithms: the ‘Clinical Face Phenotype Space’ [15] and ‘OpenFace’ [16] and has already demonstrated its use to distinguish KdVS, Schuurs-Hoeijmakers syndrome (PACS1, OMIM #615009), Jansen-de Vries syndrome (PPM1D, OMIM #617450) and Chung-Jansen syndrome (PHIP, OMIM #617991) from an age, gender and ethnicity matched control cohort of individuals with intellectual disability when comparing their 2D photographs [14].
In this study, we tested the ability of this hybrid model to differentiate between the molecular subtypes of KdVS and to differentiate between two ID syndromes, KdVS and 22q11.2DS. The latter was specifically chosen as it shows similarity to KdVS in especially the facial features. In addition, we determined whether our tool allows to diagnose patients with a KANSL1 variant of unknown clinical significance by exhibiting significant similarity in facial phenotype to patients diagnosed with KdVS or matched controls.
Subjects and methods
Data collection
For this study, two test sets were collected, each containing 2D photographs of Caucasian individuals with a confirmed clinical and molecular diagnosis of either KdVS or 22q11.2DS. All 2D facial photographs were taken from an approximately frontal position under uncontrolled conditions. The individual 2D photographs used in this study consist of previously published patients (n = 78) [4, 5, 7, 17,18,19] and non-published patients (n = 67).
The KdVS test set consisted of 97 2D photographs, of which 78 patients were previously diagnosed with a (de novo) 17q21.31 microdeletion and 19 patients with a variant (likely) causing KdVS in KANSL1 (e.g. class 4 or 5 variant according to ACMG guidelines). Each patient was represented by one 2D photograph. And although 71 of these patients were used in a previous study [14], the method of analysis ensured a patient was analysed without the use of previously obtained results of other analysis, not within previous studies, nor from within the same study. The 22q11.2DS test set consisted of 48 photographs. All 22q11.2DS patients had deletions encompassing 1.5–3.0 Mb, associated with velo-cardio-facial syndrome (OMIM #188400). The age and gender distribution of the several analysed (sub)groups are shown in Table 1 and Fig. 1. It is important to mention that both age (p = 0.813 in the KdVS cohort and p = 0.477 when comparing the KdVS and 22q11.2DS cohorts) and gender (p = 0.446 and p = 0.482 respectively) did not significantly differ between all investigated patient groups, since it is known that those characteristics can influence the results.

The age distribution of our patient groups: no significant difference was observed in all.
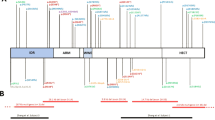
In a second phase of the project, three patients with a variant of unknown clinical significance in KANSL1 (ACMG class 3) were analysed to help establish a conclusive diagnosis using the hybrid facial tool. Of these, patient 1 had a de novo splice site variant (NM_001193466.1:c.2542-3C>A) and mRNA sequencing showed the formation of a new transcript (r.2546_2548del). This transcript, however, only missed the first three base pairs of exon 11 (exons are numbered consecutively from 1 to 15 according to NM_001193466.1), leading to an in-frame deletion of only one amino acid (p.Gln849del). The transcript was not subject to nonsense mediated decay. Since only one amino acid is deleted due to this variant, the variant was still classified as a variant of unknown significance, even after RNA sequencing. Patient 2 had a de novo missense variant (NM_001193466.1:c.1448G>A (p.(Gly483Glu))), with low level (~15%) mosaicism. Patient 3 had a missense variant (NM_015443.3:c.530A>G p.(Asn177Ser)) as well, but was adopted and therefore inheritance of the variant could not be determined.
For all three patients, our team of clinicians (DK and BBAdV) initially doubted the diagnosis of KdVS, either because of the variant and/or the clinical phenotype. To ensure accessibility, the data of these three new patients were added to the Leiden Open Variation Database (LOVD, https://databases.lovd.nl/shared/genes/KANSL1) with submission IDs #00307401, #00307402 and #00307403.
For both the KdVS and 22q11.2DS patients, 145 age, gender and ethnicity matched controls were selected. When analysing a patient with a VUS in KANSL1, an age, gender and ethnicity matched control for that specific patient was added. With the introduction of these matched controls, possible bias introduced by age, gender or ethnicity differences is overcome. These matched controls were patients with intellectual disability, seen in the outpatient clinic of the Radboud University Medical Centre, Nijmegen, the Netherlands. To ensure anonymity all photographs were converted into unidentifiable feature vectors before analysis. This study was approved by the ethics committee of the Radboudumc (number 2018-4733).
Facial recognition analyses
The 2D photographs were analysed using the hybrid model for quantitative facial phenotyping, which combines the facial dysmorphism analysis of the ‘Clinical Face Phenotype Space’ pipeline with the facial recognition system of the ‘OpenFace’ pipeline, as described by van der Donk et al. [14]. First, we compared the 2D photographs of the 22q11.2DS patients with the KdVS patients, to verify if our model is able to accurately detect small differences in the facial phenotype.
After extraction of the hybrid features for each of the 145 patient 2D photographs, the nearest neighbour (Euclidean distance) for each patient was determined. For both groups the number of patients for whom the nearest neighbour is from the same group—assigned as a match—and the number of patients for whom the nearest neighbour belongs to the other group—assigned as no match—were determined. The observed values were compared with our expected match/no match values, determined by:
in which n1 represents our first subset of 2D photographs, corresponding to the KdVS patients, and n2 to the 22q11.2DS patients.
For each outcome, observed and expected frequencies of matches were compared by calculating the exact goodness of fit test, because small numbers of 2D photographs were available for some of the test sets. Our null hypothesis stated that the number of observed matches is equal to or lower than the expected number of matches. We calculated the probability of getting a deviation from the null hypothesis larger than (or equal to) the observed result and the p value was set at 0.05.
Results
In total, the 2D photographs of 145 patients were analysed in this study using the previously described hybrid model for quantitative facial phenotyping [14].
When comparing the 22q11.2DS patients (n = 48) with all the KdVS patients (n = 97), we observed significantly more matches than to be expected by random chance: respectively 25 vs. 15.7 for the 22q11.2DS patients (p = 0.0052) and 90 vs. 64.7 for the KdVS patients (p = 7.5 × 10−10, see Table 2). This shows that the model can distinguish patients with KdVS from patients with 22q11.2DS. Performing the same analysis within the KdVS patient group and comparing patients with the 17q21.31 microdeletion (n = 78) and patients with a truncating KANSL1 variant (likely) causing KdVS (n = 19), no significant difference in observed vs. expected matches was found (Table 3).
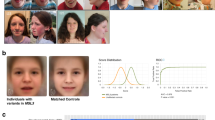
Subsequently, 2D photographs from three patients identified with a KANSL1 variant of unknown clinical significance were separately analysed (Supplementary Clinical Data; Supplementary Molecular Characteristics; Fig. 2) within the total cohort of 97 KdVS patients and their matched controls. For all three patients, clinicians initially doubted the diagnosis of KdVS, because of the phenotype and facial features. The model provided a phenotypic readout to support the interpretation of the variants of unknown clinical significance. We accomplished this by comparing the Euclidean distance of the patients with a KANSL1 variant of unknown clinical significance with their nearest KdVS patient with the Euclidean distance of the controls and their nearest KdVS patient. With this method, we checked whether the patients with a KANSL1 variant of unknown clinical significance were nearer to the controls or the KdVS patients. Statistical significance was calculated utilising the Mann–Whitney U test, p value set at 0.05. None of the three patients clustered significantly within the KdVS patient group using the nearest neighbour approach (p = 0.636, 0.144 and 0.877; Table 4).

Below: age and gender matched KdVS patients with the 17q21.31 microdeletion. Note the similarity in facial features in the patients with the microdeletion and the difference with the patients with the atypical variants. Dotted lines denote the exon boundaries: the digits correspond to the numbering of the amino acids of the KANSL1 gene.
Discussion
We have previously shown that our hybrid model for quantitative facial phenotyping can distinguish the facial phenotype of several intellectual disability syndromes, respectively Schuurs-Hoeijmakers syndrome (PACS1, OMIM #615009), Jansen-de Vries syndrome (PPM1D, OMIM #617450) and Chung-Jansen syndrome (PHIP, OMIM #617991) from an age and gender matched control group of patients with intellectual disability [14]. This model showed to be helpful in ascertaining whether a variant of unknown clinical significance in one of those genes can be regarded as (possible) pathogenic and thereby causative. In these initial analyses, the patients were compared with controls with intellectual disability with a wide range of dysmorphic features.
To further demonstrate the capabilities of the model, we here compared two more facially resembling intellectual disability syndromes: the 22q11.2DS and KdVS. Our analyses show that the model is able to detect small differences and differentiate between patients diagnosed with these two facially overlapping ID syndromes.
Within the KdVS sample our model could not detect any differences in facial features for either the 17q21.31 microdeletion or KANSL1 loss-of-function variants (likely) vausing KdVS, thereby statistically quantifying expert clinical observations that could not discriminate KdVS caused by KANSL1 variants (likely) causing KdVS or deletions, in an unbiased way [7].
Moreover, we have looked at three atypical KdVS patients with variants of unknown clinical significance in KANSL1—as the classical KdVS patients have truncating variants in KANSL1. None of the three were quantified as having the typical facial phenotype associated with KdVS: our model supported the findings of the clinicians, as none of the patients with variants of unknown clinical significance in KANSL1 clustered significantly with the KdVS patient group. It should be noted that since most of the patients in our study are of Caucasian descend, the results of this algorithm are not (yet) validated in patients with a different ethnic background.
In conclusion, we further demonstrate the capabilities of our hybrid model of quantitative facial phenotyping to differentiate in the facial phenotype of intellectual disability syndromes, even in two syndromes with overlapping facial dysmorphic features. Next to that, we provide further evidence that there is no facially significant difference in KdVS patients with either the 17q21.31 microdeletion or a truncating KANSL1 variant and that the model allows to classify variants of unknown clinical significance in the KANSL1 gene.
References
Koolen DA, Vissers LE, Pfundt R, de Leeuw N, Knight SJ, Regan R, et al. A new chromosome 17q21.31 microdeletion syndrome associated with a common inversion polymorphism. Nat Genet. 2006;38:999–1001.
Sharp AJ, Hansen S, Selzer RR, Cheng Z, Regan R, Hurst JA, et al. Discovery of previously unidentified genomic disorders from the duplication architecture of the human genome. Nat Genet. 2006;38:1038–42.
Shaw-Smith C, Pittman AM, Willatt L, Martin H, Rickman L, Gribble S, et al. Microdeletion encompassing MAPT at chromosome 17q21.3 is associated with developmental delay and learning disability. Nat Genet. 2006;38:1032–7.
Koolen DA, de Vries BBA. KANSL1-related intellectual disability syndrome. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, et al. editors. GeneReviews((R)). Seattle (WA): University of Washington, Seattle University of Washington, Seattle. GeneReviews is a registered trademark of the University of Washington, Seattle. All rights reserved.; 2013.
Koolen DA, Kramer JM, Neveling K, Nillesen WM, Moore-Barton HL, Elmslie FV, et al. Mutations in the chromatin modifier gene KANSL1 cause the 17q21.31 microdeletion syndrome. Nat Genet. 2012;44:639–41.
Zollino M, Orteschi D, Murdolo M, Lattante S, Battaglia D, Stefanini C, et al. Mutations in KANSL1 cause the 17q21.31 microdeletion syndrome phenotype. Nat Genet. 2012;44:636–8.
Koolen DA, Pfundt R, Linda K, Beunders G, Veenstra-Knol HE, Conta JH, et al. The Koolen-de Vries syndrome: a phenotypic comparison of patients with a 17q21.31 microdeletion versus a KANSL1 sequence variant. Eur J Hum Genet. 2016;24:652–9.
McDonald-McGinn DM, Sullivan KE. Chromosome 22q11.2 deletion syndrome (DiGeorge syndrome/velocardiofacial syndrome). Medicine. 2011;90:1–18.
Campbell IM, Sheppard SE, Crowley TB, McGinn DE, Bailey A, McGinn MJ, et al. What is new with 22q? An update from the 22q and You Center at the Children’s Hospital of Philadelphia. Am J Med Genet A. 2018;176:2058–69.
McDonald-McGinn DM, Sullivan KE, Marino B, Philip N, Swillen A, Vorstman JA, et al. 22q11.2 deletion syndrome. Nat Rev Dis Prim. 2015;1:15071.
Dudding-Byth T, Baxter A, Holliday EG, Hackett A, O’Donnell S, White SM, et al. Computer face-matching technology using two-dimensional photographs accurately matches the facial gestalt of unrelated individuals with the same syndromic form of intellectual disability. BMC Biotechnol. 2017;17:90.
Gurovich Y, Hanani Y, Bar O, Nadav G, Fleischer N, Gelbman D, et al. Identifying facial phenotypes of genetic disorders using deep learning. Nat Med. 2019;25:60–4.
Nellaker C, Alkuraya FS, Baynam G, Bernier RA, Bernier FPJ, Boulanger V, et al. Enabling global clinical collaborations on identifiable patient data: the Minerva initiative. Front Genet. 2019;10:611.
van der Donk R, Jansen S, Schuurs-Hoeijmakers JHM, Koolen DA, Goltstein L, Hoischen A, et al. Next-generation phenotyping using computer vision algorithms in rare genomic neurodevelopmental disorders. Genet Med. 2019;21:1719–25.
Ferry Q, Steinberg J, Webber C, FitzPatrick DR, Ponting CP, Zisserman A, et al. Diagnostically relevant facial gestalt information from ordinary photos. Elife. 2014;3:e02020.
Amos B, Ludwiczuk B, Satyanarayanan M. Openface: a general-purpose face recognition library with mobile applications. S. Technical Report, CMU-CS-16-118, CMU School of Computer Science, Pittsburgh, PA, 2016.
Dubourg C, Sanlaville D, Doco-Fenzy M, Le Caignec C, Missirian C, Jaillard S, et al. Clinical and molecular characterization of 17q21.31 microdeletion syndrome in 14 French patients with mental retardation. Eur J Med Genet. 2011;54:144–51.
Koolen DA, Sharp AJ, Hurst JA, Firth HV, Knight SJ, Goldenberg A, et al. Clinical and molecular delineation of the 17q21.31 microdeletion syndrome. J Med Genet. 2008;45:710–20.
Hoyer J, Dreweke A, Becker C, Gohring I, Thiel CT, Peippo MM, et al. Molecular karyotyping in patients with mental retardation using 100K single-nucleotide polymorphism arrays. J Med Genet. 2007;44:629–36.
Acknowledgements
We are grateful to the patients and their parents for their participation, to the Dutch Organisation for Health Research and Development: ZON-MW grants 912-12-109 (to BBAdV and LELMV) and 916-16-015 (to JYHK), Donders Junior researcher grant 2019 (BBAdV and LELMV), Aspasia grant 015.014.066 (to LELMV). Inclusion of Radboudumc data was in part supported by the Solve-RD project that has received funding from the European Union’s Horizon 2020 research and innovation programme under grant agreement no. 779257. The patient images used for analysis in this study cannot be made openly available due to patient privacy concerns. Please contact the authors with specific queries regarding data access.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Dingemans, A.J.M., Stremmelaar, D.E., van der Donk, R. et al. Quantitative facial phenotyping for Koolen-de Vries and 22q11.2 deletion syndrome. Eur J Hum Genet 29, 1418–1423 (2021). https://doi.org/10.1038/s41431-021-00824-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-021-00824-x
This article is cited by
-
PhenoScore quantifies phenotypic variation for rare genetic diseases by combining facial analysis with other clinical features using a machine-learning framework
Nature Genetics (2023)
-
Identification of KANSL1 as a novel pathogenic gene for developmental dysplasia of the hip
Journal of Molecular Medicine (2022)
-
Establishing the phenotypic spectrum of ZTTK syndrome by analysis of 52 individuals with variants in SON
European Journal of Human Genetics (2022)
