
Abstract
The BCAP31 gene, located at Xq28, encodes BAP31, which plays a role in ER-to-Golgi anterograde transport. To date, BCAP31 pathogenic variants have been reported in 12 male cases from seven families (six loss of function (LoF) and one missense). Patients had severe intellectual disability (ID), dystonia, deafness, and central hypomyelination, delineating a so-called deafness, dystonia and cerebral hypomyelination syndrome (DDCH). Female carriers are mostly asymptomatic but may present with deafness. BCAP31 is flanked by the SLC6A8 and ABCD1 genes. Contiguous deletions of BCAP31 and ABCD1 and/or SLC6A8 have been described in 12 patients. Patients with deletions including BCAP31 and SLC6A8 have the same phenotype as BCAP31 patients. Patients with deletions of BCAP31 and ABCD1 have contiguous ABCD1 and DXS1375E/BCAP31 deletion syndrome (CADDS), and demonstrate a more severe neurological phenotype with cholestatic liver disease and early death. We report 17 novel families, 14 with intragenic BCAP31 variants (LoF and missense) and three with a deletion of BCAP31 and adjacent genes (comprising two CADDS patients, one male and one symptomatic female). Our study confirms the phenotype reported in males with intragenic LoF variants and shows that males with missense variants exhibit a milder phenotype. Most patients with a LoF pathogenic BCAP31 variant have permanent or transient liver enzyme elevation. We further demonstrate that carrier females (n = 10) may have a phenotype comprising LD, ID, and/or deafness. The male with CADDS had a severe neurological phenotype, but no cholestatic liver disease, and the symptomatic female had moderate ID and cholestatic liver disease.
Similar content being viewed by others


Introduction
The BCAP31 gene (MIM 300398) is located at Xq28 and encodes B-cell-receptor-associated protein 31 (BAP31), a ubiquitous 31-kDa chaperone protein highly expressed in neurons [1]. It is the most abundant of endoplasmic reticulum (ER) membrane proteins [2] and plays a role in regulation of apoptosis, protein transport, and degradation. BAP31 has a role in the export of secreted proteins [3, 4], and their targeting to the ER-associated-degradation pathway [5, 6]. BAP31 also serves as a cargo receptor for the export of transmembrane proteins. [1].
In 2013, Cacciagli et al. [7] described a specific phenotype associated with BCAP31 loss-of-function (LoF) anomalies in seven males from three families with severe to profound developmental delay (DD) or intellectual disability (ID), dystonia, seizures, sensorineural hearing loss (SNHL), and central myelination delay, which defined the deafness, dystonia, and cerebral hypomyelination syndrome (for DDCH, MIM 300475). In all instances, the pathogenic BCAP31 variants were inherited from asymptomatic mothers. Functional studies were undertaken on patient fibroblasts and supported the evidence that BAP31 plays a role in ER-to-Golgi exchanges. No evidence of accumulation of misfolded proteins or activation of either UPR or cell-death programs was found. It was hypothesized that the key role of ER protein trafficking in the myelination process [8] could explain the white matter abnormalities.
Four additional male patients from three families with BCAP31 LoF anomalies and a similar phenotype were subsequently reported [9,10,11]. Vittal et al reported two affected brothers (twins) with a 6 bp deletion in c.261_266delGCTTCT (c.60_65delGCTTCT according to reference transcript NM_001139441.1) resulting in a protein change of p.(Leu87_Leu89delinsPhe) (p.(Leu20_Leu22delinsPhe) according to the protein produced by transcript NM_001139441.1) that can be considered as a missense variant [12]. The phenotype of one of the two brothers is milder than in previously described patients with BCAP31 LoF variants, as he acquired partial language and basic academic skills.
Liver enzymes were elevated in half of the reported patients, either permanently or intermittently, sometimes during febrile episodes. Patients with pathogenic BCAP31 variants/intragenic deletions did not display cholestasis or hepatic failure, except one with acute liver cytolysis and cholestasis concomitant to an episode of intestinal necrosis [11]. All female carriers reported to date were asymptomatic, except one with isolated SNHL [9].
BCAP31 is flanked by SLC6A8 (MIM 300036) on its centromeric side, and ABCD1 (MIM 300371) on its telomeric side. Contiguous deletions of BCAP31 and ABCD1 and/or SLC6A8 have been described. Pathogenic ABCD1 LoF variants are responsible of X-linked adrenoleukodystrophy, a neurodegenerative condition that affects the central nervous system white matter and the adrenal cortex, that can reveal itself in childhood (cerebral form) or adulthood (adrenomyeloneuropathy) [13]. The childhood onset of the disease is characterized by progressive impairment of cognition, behavior, vision, hearing, and motor function. However, neurodegeneration does not start before 2.7 years. Pathogenic SLC6A8 LoF variants result in cerebral creatine deficiency and the patients display mild to severe ID, seizures and behavioral problems [14]. SNHL, dystonia and chorea are rare. Patients with deletions of BCAP31 and ABCD1 have Contiguous ABCD1 and DXS1375E (BCAP31) Deletion Syndrome (CADDS, MIM 300475) [15]. The six male patients described with CADDS had common signs with DDCH, such as severe DD, dystonia, white matter abnormalities and deafness. However, all of these six patients had chronic cholestatic liver disease, and a more severe course of the disease, as all died in the first year of life [15,16,17,18,19]. The only symptomatic female with a deletion encompassing both BCAP31 and ABCD1 reported to date is a 9-year-old girl with severe ID, autism spectrum disorder, microcephaly and deafness, but without white matter anomalies or dystonia [20].
Six patients with a deletion including BCAP31 and SLC6A8 have been described (including a family of four patients described by Cacciagli et al.) [7, 18, 21]. Their phenotype was more severe than most patients with pathogenic SLC6A8 variants, and they had symptoms and signs similar to BCAP31 patients, such as dystonia, choreoathetosis, SNHL and white matter abnormalities, which are unusual in cerebral creatine deficiency. Thus, most of their phenotype was ascribed to the deletion of BCAP31 [7].
We assessed 17 novel families with BCAP31 anomalies, 14 families with intragenic LoF or missense variants (16 males and 9 symptomatic females) and three families with a deletion of BCAP31 and adjacent genes (comprising two CADDS patients, one male and a symptomatic female). The symptomatic female carriers had SNHL and/or DD/ID. We also describe two asymptomatic female carriers with somatic mosaicism which is particularly relevant for genetic counseling.
Patients and methods
Male and female individuals with BCAP31 variants and deletions were recruited from different cohorts through national and international collaborations. For two families, the patients were part of a cohort of cerebral palsy patients.
Genetic testing was performed by chromosomal microarray (n = 3 deletions), whole-genome sequencing (n = 1), whole-exome sequencing (n = 8), TrueSight One gene panel (n = 1), X chromosome sequencing (n = 1), targeted gene panel for ID (n = 1), targeted gene panel for mitochondrial diseases (n = 1), or direct Sanger sequencing of BCAP31 (n = 1).
Variants were annotated using NM_001139441.1 as a reference sequence and were classified according to the ACMG criteria [22, 23]. Exons are numbered according to Cacciagli et al. [7] and correspond to transcript ENST00000458587.8.
Intragenic variants were submitted to the gene variant database LOVD at https://databases.lovd.nl/shared/individuals/BCAP31 (Patients 1–11: individuals 00315922–00315932, Patient 12: individual 00317967, Patient 13: individual 00315934, Patient 14: individual 00314857, Patients 15–18: individuals 00314696–00314699). Large deletions including BCAP31 and adjacent genes were submitted to ClinVar at https://www.ncbi.nlm.nih.gov/clinvar (Patients 19–21: individuals SCV001450740–SCV001450742).
Each patient’s referring physician filled out a table with detailed general data, family history, pregnancy and labor, neonatal period, developmental milestones, neurological signs, behavioral and epilepsy history, and other detailed clinical history or features (senses, liver, cardiac, and respiratory). When possible, pictures and/or videos of the patients and brain MRI were reviewed.
Parental written informed consent was obtained for all affected patients. Genetic testing was performed in accordance with the respective national ethics guidelines and approved by the local authorities in the participating study centers.
Results
Male patients with intragenic variants of BCAP31
Male patients with LoF variants
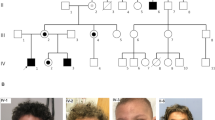
A total of 12 males from 11 families had intragenic LoF variants (Table 1, P1–P12 Families 1–11, and Fig. 1), that were all maternally inherited except one that was de novo. Somatic mosaicism was found in the mother of P12.

The variants identified in patients of our study are located above the gene, and those previously reported are underneath the gene. Reference transcript: NM_001139441.1. Exons are numbered according to Cacciagli et al. [7] and correspond to transcript ENST00000458587.8. Full splice variant denomination is NC_000023.11(NM_001139441.1):g.153723152C>T for P2 and P3; and NC_000023.11(NM_001139441.1):g.153702006C>T for P8.
The median age of these patients at last examination was 9 years 5 months (2.4–28 years), and all but one were alive at the time of the study. All patients presented with severe to profound DD, none achieved walking, all had absent or limited speech, and all but one had limited or no purposeful use of hands. Neurological examination showed dystonic postures and/or choreic movements (12/12), microcephaly (7/11), and increased muscle tone/pyramidal signs (6/11). The diagnosis of cerebral palsy was initially considered in P5. Seizures were seen in 3/12 patients (petit mal for one, not detailed in the 2 others). The disease appeared to be progressive for two patients. Brain MRI was abnormal in 9/11 patients. White matter (WM) anomalies were the most frequent (7/11), and were described as reduced WM volume, myelination delay, hypomyelination, and WM hyperintensities. These anomalies disappeared with time in one patient at the age of 12 years. Other reported anomalies were cortical atrophy (4/11), thin corpus callosum (3/9), atrophy of basal ganglia and thalami (2/11), and hypoplastic cerebellar vermis (2/11).
Moderate to profound SNHL was seen in 9/10 (with normal inner ear CT scan for one patient) and strabismus was frequent (6/11). Small stature (−2 SD or below) was seen in 7/12 patients, with weight on −2 SD or below in 6/12. Some common facial features were noted such as hypotonic face and deep-set eyes. Unexplained fever was reported in two patients. No major cardio-respiratory anomalies were reported.
Liver enzymes were increased in 8/10, either transiently (n = 4) or permanently (n = 4). Two patients (P7 and P12) underwent liver biopsy, that showed mild-to-moderate mononuclear portal tract inflammatory infiltrate consistent with mild chronic hepatitis in P7, and mitochondrial inclusions with a regular/periodic pattern in P12. The latter finding could represent abnormalities of cristae and gave an overall morphology of crystalline inclusions. Mitochondrial complex activities were normal in the fibroblast of two brothers (P2 and P3). CSF neurotransmitters showed mild cerebral folate deficiency (5-MTHF 30 nM, normal 40–187 nM) in one patient (P10). He was treated with oral folinic acid with minimal clinical effect.
Detailed molecular results of these patients are presented in Fig. 1. Interestingly, the variant c.365_366del was found in unrelated patients P5 and P10.
Male patients with missense variants
Four male patients from two families had missense variants (Table 2, P14–P17, Families 13 and 14, and Fig. 1). Median age at last examination was 19.25 years (3–36 years) and all but one were alive at the time of the study.
The patients had mild (1/4), moderate (1/4), or severe (2/4) DD/ID. Unsupported sitting was achieved in 3/3, assisted walking for 2/3, all had partial language skills (words for three, sentences for one), and three patients attended a special needs school, one was reported to be able to read simple texts. Dystonic postures and/or choreic movements were seen in all patients and two had increased muscle tone/pyramidal signs. The diagnosis of cerebral palsy was initially considered in these patients. The use of hands was reported to be purposeful for one. Microcephaly was seen in 1/3. None of these patients had seizures or apparent progressive course of the disease. For P14, brain MRI was normal at 14 months.
SNHL was seen in P14 with normal inner ear CT scan, and conductive hearing loss was found in P15. No ophthalmological data were obtained, and no liver dysfunction was reported. Two patients had persistent failure to thrive. P14 had a hypotonic face. His supposedly affected uncle had deep-set eyes with high and narrow nasal bridge, similar to other DDCH patients. No specific facial features were noted for the patients of family 14.
In P14 (Family 13), the NM_001139441.1:c.47T>A; p.(Val16Asp) missense variant affects a highly conserved amino-acid within the B-cell receptor-associated 31-like domain, is not reported in gnomAD and is estimated disease causing by pathogenicity predictors. This variant is not reported in gnomAD.
In P15 (Family 14), X chromosome exome sequencing identified 16 variants in total, including the BCAP31 variant Hg38: chrX:153715545G>T, NM_001139441.1:c.338C>A. After in silico analysis, all variants except BCAP31 were not flagged for follow up. This variant affects a moderately conserved serine residue located in the BAP31 superfamily domain. It is not present in gnomAD and has a CADD (phred) of 23.5. Preliminary data in P18, a symptomatic female (direct cousin of P15), suggests that her fibroblasts have larger/more swollen ER than controls, and abnormal golgi staining (data not shown). The most substantial evidence for the pathogenicity of the variant in this family is its presence in all symptomatic individuals and carrier mothers. However, according to ACMG criteria, these two missense variants remain classified as VUS to date.
Symptomatic female carriers of intragenic BCAP31 anomalies
Females with LoF variants
Three carrier mothers were symptomatic, two with SNHL (family 2 and 4), and one with apparent mild ID (family 3).
P13 was the only affected individual in family 12 and had severe ID, achieved sitting at 18 months, walking at 4 years, and had limited speech at 7 years. She had microcephaly and lower limb spasticity, but no dystonia, chorea, or seizures. She had hearing loss and persistent strabismus. She had deep-set eyes with high and narrow nasal bridge. Brain MRI at 3 years old showed thin corpus callosum, enlarged cisterna magna, enlarged subarachnoid frontal space but no white matter anomalies. Liver function was normal. X-inactivation studies in peripheral blood showed significant bias at the FRAXA locus (93/7%) and 88/12% at the HUMARA locus. Her mother was found to have a somatic mosaicism for the pathogenic BCAP31 variant (3%).
Females with missense variants
In Family 13, the four confirmed female carriers had isolated SNHL. X-inactivation chromosome studies in peripheral blood were inconclusive, showing moderate skewing in one (90/10), no skewing for the second, and uninformative results in the third.
In Family 14, a carrier female (Table 2, P18) had global DD, acquired walking at age 17 months and had language delay. She had mild ID, attended a special needs school and could read. She had mild ataxic gait and drop attacks, but no dystonia or chorea. Mild conductive hearing loss was noted in infancy as well as atypical retinal pigmentation. No specific facial features were noted. No data were obtained for liver function. X-inactivation was random in peripheral blood, but skewed toward the variant allele in cultured fibroblasts, as sequencing of the BCAP31 transcript from fibroblast cDNA, showed only presence of the variant transcript. Functional studies were undertaken on the fibroblasts showing mildly altered ER and Golgi as described by Cacciagli et al. but were not statistically significant (data not shown).
Patients with deletion of BCAP31 and adjacent genes (Table 3 and Fig. 2)
Male patient with non-CADDS deletion
P19 (Family 15), had a de novo deletion including BCAP31, SLC6A8, DUSP9, and PNCK (NC_000023.10:g.(152886255_152976269)del). He had profound DD, infantile spasms, pyramidal signs but no dystonia or chorea. Brain MRI at 2 years old showed abnormal WM and global atrophy. SNHL was diagnosed at birth. Growth was normal including OFC. He had no cholestatic liver disease, however developed reversible acute liver failure during a lung infection that lead to sepsis.

On the top, non-CADDS patients, with deletions including BCAP31 and adjacent genes in 5′ (excluding ABCD1). On the bottom, CADDS patients with deletions including BCAP31 and ABCD1. Reference transcript: NM_001139441.1. Exons are numbered according to Cacciagli et al. [7] and correspond to transcript ENST00000458587.8.
Male patient with CADDS
P20 (Family 16) had a de novo 60-kb deletion including BCAP31, ABCD1, and PLXNB3 (NC_000023.10:g.(152982350_153041544)del). He had severe DD with no acquired milestones at 16 months, choreic movements, and frequent opisthotonus. Brain MRI at 1 and 10 months showed thalamic hyperintensities with normal WM. He had permanent moderate liver enzyme elevation, but no cholestasis. He had normal hearing, bilateral strabismus, episodes of unexplained fever, and recurrent respiratory infections. He was born with severe IUGR followed by severe growth impairment. He was treated for adrenal and exocrine pancreatic deficiency. Lung CT was undertaken at 15 months because of chronic hypoxia and showed unexplained interstitial lung infiltrate. He died of respiratory failure at 16 months.
Symptomatic female with a deletion of BCAP31 and ABCD1
P21 (Family 17) had a deletion including SLC6A8, BCAP31, exon 1 of ABCD1, DUSP9, PCNK (NC_000023.10:g.(152882907_152991027)del). She was adopted and parental analysis could not be performed. She had moderate ID, walked at 24 months and said a few words at 3 years. She had no seizures and no microcephaly. Brain MRI at 15 months showed diffuse WM abnormalities predominant in periventricular region and global atrophy. Spectroscopy showed reduced creatine peak at 50% due to SLC6A8 deletion. She also had a SNHL, strabismus, and hypermetropia. Liver dysfunction was reported with chronic cholestasis, moderate hepatic failure, transient episodes of liver enzyme elevation, and liver biopsy showed signs of cholangiopathy.
Discussion
Males with intragenic pathogenic BCAP31 variants
The phenotype of the 12 males of this study with intragenic LoF variants is similar to that of previously described cases, hereby confirming a homogeneous clinical involvement in all reported cases to date. The pathogenic BCAP31 variants were inherited in ten families, and one mother displayed somatic mosaicism (patient 12). All male patients displayed severe DD/ID with no walking, absent, or very limited language skills and dystonia or chorea. Microcephaly and increased muscle tone/pyramidal signs were frequent. Seizures were present in few individuals and two patients had an apparently progressive course of disease, which has not yet been reported. Common facial features were noted in some patients with hypotonic face and deep-set eyes with narrow and high nasal bridge, the latter seemed to be more obvious with time (Fig. 3). Physicians considered the diagnosis of cerebral palsy in P5 (LoF variant), in individuals of Family 14 (missense variant) and in a presumably affected relative of P14. Hence, it would be relevant to search for pathogenic BCAP31 variants in patients presenting with unexplained cerebral palsy. As in previous patients, brain MRI frequently showed abnormal WM (Fig. 4). P2 showed surprising results over time, with myelination delay at 9 months, WM hyperintensities at 10 years, and normalized WM at 12 years old. It would be interesting to repeat the MRI in other patients to further characterize WM changes during the course of the disease. Other signs were reported but inconsistent, including cortical atrophy, thin corpus callosum, basal ganglia anomalies (atrophic, small, hyperintense on T2-weighted MRI sequences), and hypoplastic cerebellar vermis. SNHL was a frequent feature, and the inner ear was radiologically normal in two patients, it would be interesting to confirm this observation in additional patients.

The males P6 and P9 have a LoF variant of BCAP31. P14 has a missense variant. P13 is a female with a LoF variant. There does not seem to be a specific facial gestalt for BCAP31 anomalies. Some common features have been noted with deep-set eyes, as P6 and P13, hypotonic face as seen in P6, P9, and P14, high nasal bridge for P6, high and narrow bridge for P9 and P13.

(A) Patient 1 (LoF intragenic pathogenic variant). At 2 years old (A1: axial FLAIR, A2–A4: axial T2): signal hyperintensity in the peritrigonal white matter (see arrows in A1), small thalami (see arrows in A3), small cerebral peduncles (see arrows in A2), paired T2 hyperintensities in the dorsal pons (see arrows in A4). (B) Patient 11 (LoF intragenic pathogenic variant). At 8 months old (B1: sagittal T1, B2: axial FLAIR): markedly decreased white matter as evidenced by a thin corpus callosum (B1 and B2) as well as the sylvian fissures nearly abutting the lateral ventricles (see arrows in B2). (C) Patient 19 (deletion of BCAP31 and adjacent genes). At 6 months (C1: sagittal T1, C2: axial T2): decreased white matter, myelination delay, cortical atrophy, thin corpus callosum and ventricular dilatation. At 2 years old (C3: sagittal T2, C4: axial T2): increased white matter anomalies, marked cortical atrophy, vermian atrophy, and ventricular dilatation.
As for molecular results, we report the first recurrent pathogenic LoF variant to our knowledge (c.365_366del). We report two further families with missense variants and suggestive clinical presentation, but that remain classified as VUS. The ID in the four males of these families was less severe than in LoF patients, similar to the patients described by Vittal et al. [12], thus a milder functional effect of the missense variants could be speculated to explain the milder ID. However, more patients and functional assessment of these variants are needed to support their involvement in the phenotype.
Symptomatic female carriers of intragenic BCAP31 variants
Our results substantiate the findings of Albanyan et al. [9] who described a carrier female with SNHL. Six females in our study also had SNHL, two with LoF variants, and four with missense variants. Interestingly, Rosenberg et al. [24] identified a patient with non-syndromic hearing loss (the sex of the patient was not mentioned) harboring a de novo duplication including BCAP31 and SLC6A8. Altogether, these data suggest that BCAP31 anomalies should be considered in females with non-syndromic hearing loss. It would be relevant for genetic counseling for female carriers, to consider the risk of having a severely affected male. Sensorineural hearing loss (SNHL) being the most common congenital sensory deficit, with an estimated prevalence of 2–3 cases per 1000 individuals it seems important to rule out other causes, genetic or not, before confirming the implication of BCAP31. Approximately half of SNHL in children is due to genetic causes, with 70% being non-syndromic. Variations in the GJB2 gene is the most common cause of non-syndromic genetic SNHL in many populations; however, there is high genetic and allelic heterogeneity, with over 100 implicated genes, which can be recessive, dominant, X-linked, or mitochondrial [25, 26].
We also report the first female carriers with mild (two carrier mothers) or severe ID (one proband). We have limited data describing the brain MRI in females, and further data are necessary to speculate about WM abnormalities in these cases. No specific facial features were noted in female carriers except for patient 13 who had severe ID and displayed deep-set eyes and narrow nasal bridge.
Patients with deletion of BCAP31 and adjacent genes (CADDS and non-CADDS)
The phenotype of the male patient of this study with a non-CADDS deletion including BCAP31 and SLC6A8 was similar to patients with a pathogenic intragenic LoF BCAP31 variant, and more severe than patients with SLC6A8 deficiency, supporting the evidence that BCAP31 is the major gene responsible for the phenotype, as discussed by previous authors [7, 19].
The phenotype of the male patient of our study with CADDS is consistent with previously published cases, with severe DD, dystonia, choreic movements, and early death. Unlike other CADDS patients, WM was normal; however, thalamic hyperintensities were noted, and hearing was normal. Unreported signs were seen in our patient, such as unexplained lung interstitial infiltrate and exocrine pancreatic deficiency.
Finally, we report the second female with a deletion of ABCD1 and BCAP31. Both patients had ID, moderate in this study and severe with ASD for the patient described by Firouzabadi et al. [20]. Both had SNHL. Neither had dystonia or chorea. The patient in this study also had cholestatic liver disease and WM abnormalities, as in male CADDS patients.
The large deletions in the three patients of this study also involve adjacent genes, as in previously reported patients. It is possible that other genes deleted in our patients and those of the literature, such as PNCK, DUSP9, PLXNB3, could also contribute to the phenotype; however, no data are available to support or exclude this hypothesis to date.
Liver phenotype in BCAP31 deficiency
Patients with intragenic BCAP31 anomalies have frequent liver enzyme elevation, either permanently or intermittently, sometimes during febrile episodes. P20 (non-CADDS deletion) displayed a unique episode of acute hepatic failure during valproic acid treatment. BCAP31 has been shown to play a role in lipid metabolism in the liver. Alteration of BCAP31 in mice leads to elevated lipid storage and subsequent inflammation [27, 28]. Interestingly, two patients with LoF BCAP31 variants displayed anomalies on liver biopsy, with liver inflammation for P7, and mitochondrial inclusions suggesting a mitochondrial disorder for P12. Shimizu et al. [11] and Albanyan et al. [9] had already suggested that BCAP31 deficiency shared similarities with mitochondrial disorders, regarding their patients who presented with bilateral hyperintensities of globus pallidus and mitochondrial anomalies on muscle biopsy [9] or complex I deficit in fibroblasts [11]. Further exploration in patients with BCAP31 deficiency would be of interest to understand the physiopathology, especially regarding the liver, and to search for signs of mitochondrial dysfunction.
No patients with BCAP31 intragenic anomalies or non-CADDS deletions had chronic cholestatic liver disease, as described in the patients with CADDS. This cholestatic disease is suggested to be linked to the deletion of both BCAP31 and ABCD1 with a supposed synergistic effect; however, there is no explanation of the mechanism up to date. The male patient in our study with CADDS had no cholestatic disease at 16 months, but had permanent moderate liver enzyme elevation. Liver biopsy in P21, the affected female CADDS carrier, showed signs of cholangiopathy.
Further data are needed to understand the potential liver dysfunction in BCAP31 deficiency and CADDS patients. In the meantime, we suggest monitoring liver function carefully in BCAP31 patients and it is suggested to administer with caution potential hepatotoxic drugs. Valproic acid should be avoided in BCAP31 patients regarding the liver failure in one of our patients. No complication has been reported for acetaminophen to date, and patient 20 frequently received acetaminophen for unexplained fever with no evident change of his liver function; however, careful monitoring could be suggested.
In summary, we report 17 novel families with pathogenic BCAP31 variants, including 14 families with LoF variants, two families with missense variants and three families with large deletions (two with CADDS and one non-CADDS). We confirm the phenotype of BCAP31 deficiency in males, with a possible milder effect of missense variants. We describe symptomatic female carriers with SNHL and/or ID, and more studies are needed to further delineate their phenotype. In most-affected male patients, the variant is inherited from the mother, and the description of two asymptomatic carrier females with somatic mosaicism prompts to give careful genetic counseling concerning the recurrence risk.
References
Manley HA, Lennon VA. Endoplasmic reticulum membrane-sorting protein of lymphocytes (BAP31) is highly expressed in neurons and discrete endocrine cells. J Histochem Cytochem J Histochem Soc. 2001;49:1235–43.
Bell AW, Ward MA, Blackstock WP, Freeman HN, Choudhary JS, Lewis AP, et al. Proteomics characterization of abundant Golgi membrane proteins. J Biol Chem. 2001;276:5152–65.
Annaert WG, Becker B, Kistner U, Reth M, Jahn R. Export of cellubrevin from the endoplasmic reticulum is controlled by BAP31. J Cell Biol. 1997;139:1397–410.
Paquet M-E, Cohen-Doyle M, Shore GC, Williams DB. Bap29/31 influences the intracellular traffic of MHC class I molecules. J Immunol. 2004;172:7548–55.
Geiger R, Andritschke D, Friebe S, Herzog F, Luisoni S, Heger T, et al. BAP31 and BiP are essential for dislocation of SV40 from the endoplasmic reticulum to the cytosol. Nat Cell Biol. 2011;13:1305–14.
Wakana Y, Takai S, Nakajima K, Tani K, Yamamoto A, Watson P, et al. Bap31 is an itinerant protein that moves between the peripheral endoplasmic reticulum (ER) and a Juxtanuclear compartment related to ER-associated degradation. Mol Biol Cell. 2008;19:1825–36.
Cacciagli P, Sutera-Sardo J, Borges-Correia A, Roux J-C, Dorboz I, Desvignes J-P, et al. Mutations in BCAP31 cause a severe X-linked phenotype with deafness, dystonia, and central hypomyelination and disorganize the Golgi apparatus. Am J Hum Genet. 2013;93:579–86.
Roboti P, Swanton E, High S. Differences in endoplasmic-reticulum quality control determine the cellular response to disease-associated mutants of proteolipid protein. J Cell Sci. 2009;122:3942–53.
Albanyan S, Al Teneiji A, Monfared N, Mercimek-Mahmutoglu S. BCAP31-associated encephalopathy and complex movement disorder mimicking mitochondrial encephalopathy. Am J Med Genet A. 2017;173:1640–3.
Rinaldi B, Van Hoof E, Corveleyn A, Van Cauter A, de Ravel T. BCAP31-related syndrome: the first de novo report. Eur J Med Genet. 2019;63:103732.
Shimizu K, Oba D, Nambu R, Tanaka M, Oguma E, Murayama K, et al. Possible mitochondrial dysfunction in a patient with deafness, dystonia, and cerebral hypomyelination (DDCH) due to BCAP31 Mutation. Mol Genet Genom Med. 2020;8:e1129.
Vittal P, Hall DA, Dames S, Mao R, Berry-Kravis E. BCAP31 mutation causing a syndrome of congenital dystonia, facial dysorphism and central hypomyelination discovered using exome sequencing. Mov Disord Clin Pract. 2016;3:197–9.
Engelen M, Kemp S, de Visser M, van Geel BM, Wanders RJ, Aubourg P, et al. X-linked adrenoleukodystrophy (X-ALD): clinical presentation and guidelines for diagnosis, follow-up and management. Orphanet J Rare Dis. 2012;7:51.
van de Kamp JM, Betsalel OT, Mercimek-Mahmutoglu S, Abulhoul L, Grünewald S, Anselm I, et al. Phenotype and genotype in 101 males with X-linked creatine transporter deficiency. J Med Genet. 2013;50:463–72.
Corzo D, Gibson W, Johnson K, Mitchell G, LePage G, Cox GF, et al. Contiguous deletion of the X-linked adrenoleukodystrophy gene (ABCD1) and DXS1357E: a novel neonatal phenotype similar to peroxisomal biogenesis disorders. Am J Hum Genet. 2002;70:1520–31.
Calhoun ARUL, Raymond GV. Distal Xq28 microdeletions: clarification of the spectrum of contiguous gene deletions involving ABCD1, BCAP31, and SLC6A8 with a new case and review of the literature. Am J Med Genet A. 2014;164A:2613–7.
Iwasa M, Yamagata T, Mizuguchi M, Itoh M, Matsumoto A, Hironaka M, et al. Contiguous ABCD1 DXS1357E deletion syndrome: report of an autopsy case. Neuropathol J Jpn Soc Neuropathol. 2013;33:292–8.
Osaka H, Takagi A, Tsuyusaki Y, Wada T, Iai M, Yamashita S, et al. Contiguous deletion of SLC6A8 and BAP31 in a patient with severe dystonia and sensorineural deafness. Mol Genet Metab. 2012;106:43–7.
van de Kamp JM, Errami A, Howidi M, Anselm I, Winter S, Phalin-Roque J, et al. Genotype-phenotype correlation of contiguous gene deletions of SLC6A8, BCAP31 and ABCD1. Clin Genet. 2015;87:141–7.
Firouzabadi SG, Kariminejad R, Vameghi R, Darvish H, Ghaedi H, Banihashemi S, et al. Copy number variants in patients with autism and additional clinical features: report of VIPR2 duplication and a novel microduplication syndrome. Mol Neurobiol. 2017;54:7019–27.
Anselm IM, Alkuraya FS, Salomons GS, Jakobs C, Fulton AB, Mazumdar M, et al. X-linked creatine transporter defect: A report on two unrelated boys with a severe clinical phenotype. J Inherit Metab Dis. 2006;29:214–9.
Nykamp K, Anderson M, Powers M, Garcia J, Herrera B, Ho Y-Y, et al. Sherloc: a comprehensive refinement of the ACMG-AMP variant classification criteria. Genet Med J Am Coll Med Genet. 2017;19:1105–17.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med J Am Coll Med Genet. 2015;17:405–24.
Rosenberg C, Freitas ÉL, Uehara DT, Auricchio MTBM, Costa SS, Oiticica J, et al. Genomic copy number alterations in non-syndromic hearing loss. Clin Genet. 2016;89:473–7.
Alford RL, Arnos KS, Fox M, Lin JW, Palmer CG, Pandya A, et al. American College of Medical Genetics and Genomics guideline for the clinical evaluation and etiologic diagnosis of hearing loss. Genet Med J Am Coll Med Genet. 2014;16:347–55.
Oza AM, DiStefano MT, Hemphill SE, Cushman BJ, Grant AR, Siegert RK, et al. Expert specification of the ACMG/AMP variant interpretation guidelines for genetic hearing loss. Hum Mutat. 2018;39:1593–613.
Wu Z, Yang F, Jiang S, Sun X, Xu J. Induction of liver steatosis in BAP31-deficient mice burdened with tunicamycin-induced endoplasmic reticulum stress. Int J Mol Sci. 2018;19:2291.
Xu J-L, Li L-Y, Wang Y-Q, Li Y-Q, Shan M, Sun S-Z, et al. Hepatocyte-specific deletion of BAP31 promotes SREBP1C activation, promotes hepatic lipid accumulation, and worsens IR in mice. J Lipid Res. 2018;59:35–47.
Acknowledgements
HC is an employee of GeneDx, Inc. JG was supported by NHMRC Grants APP1155224 and APP1091593 and Channel 7 Children’s Research Foundation. AS’s research reported in this publication was supported by the National Human Genome Research Institute of the National Institutes of Health under Award Number U01HG009599. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. Amendola et. al. American Journal of Human Genetics. 2018 Sep 6;103(3):319–327. doi: 10.1016/j.ajhg.2018.08.007.
Care4Rare Canada Consortium
Kym Boycott34, Michael Brudno35, Francois Bernier36, Clara van Karnebeek37, David Dyment38, Kristin Kernohan39, Micheil Innes36, Ryan Lamont36, Jillian Parboosingh36, Deborah Marshall36, Christian Marshall35, Roberto Mendoza35, James Dowling35, Robin Hayeems35, Bartha Knoppers40, Anna Lehman37, Sara Mostafavi37
Author information
Authors and Affiliations
Consortia
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Members of the Care4Rare Canada Consortium are listed below Acknowledgements.
Supplementary information
Rights and permissions
About this article

Cite this article
Whalen, S., Shaw, M., Mignot, C. et al. Further delineation of BCAP31-linked intellectual disability: description of 17 new families with LoF and missense variants. Eur J Hum Genet 29, 1405–1417 (2021). https://doi.org/10.1038/s41431-021-00821-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-021-00821-0
