
Abstract
Dutch genome diagnostic centers (GDC) use next-generation sequencing (NGS)-based diagnostic applications for the diagnosis of primary immunodeficiencies (PIDs). The interpretation of genetic variants in many PIDs is complicated because of the phenotypic and genetic heterogeneity. To analyze uniformity of variant filtering, interpretation, and reporting in NGS-based diagnostics for PID, an external quality assessment was performed. Four main Dutch GDCs participated in the quality assessment. Unannotated variant call format (VCF) files of two PID patient analyses per laboratory were distributed among the four GDCs, analyzed, and interpreted (eight analyses in total). Variants that would be reported to the clinician and/or advised for further investigation were compared between the centers. A survey measuring the experiences of clinical laboratory geneticists was part of the study. Analysis of samples with confirmed diagnoses showed that all centers reported at least the variants classified as likely pathogenic (LP) or pathogenic (P) variants in all samples, except for variants in two genes (PSTPIP1 and BTK). The absence of clinical information complicated correct classification of variants. In this external quality assessment, the final interpretation and conclusions of the genetic analyses were uniform among the four participating genetic centers. Clinical and immunological data provided by a medical specialist are required to be able to draw proper conclusions from genetic data.
Similar content being viewed by others
Introduction
Application of next-generation sequencing (NGS) has become common practice in clinical laboratories for the genetic diagnosis of multiple inherited disorders [1]. In the Netherlands, all genome diagnostic centers (GDC) have implemented one or more NGS-based diagnostic applications [2]. These diagnostic methods currently are the preferred method for genetic diagnostic analysis of primary immunodeficiency disorders (PID) [3]. NGS testing can be described by multiple stages of analysis [4]. The primary stage involves converting images or signals from the sequencing instrument into sequence reads, followed by read mapping in the second stage. During the second stage, data are further processed with the aim to generate a file in which sequence variations with high-level summaries and annotations are stored. This file has a standardized format named variant call format (VCF) [5]. Data generated during this process enter the tertiary stage.
The aim of variant interpretation, or tertiary stage, is to link sequence variants to phenotypic features of the patient, and thus provide a potential diagnosis. During this stage, sequence variants from unannotated VCF files are interpreted according to ACMG guidelines [6]. Clinically, important findings are identified to generate a final report for the medical specialist. Many laboratories have developed several tools to filter out specific known benign variants so that only potential detrimental variants need to be assessed by the clinical laboratory geneticists [7, 8].
The generation of large amounts of data and the use of complex bioinformatic pipelines to analyze these data and classify variants also cause various challenges. The bioinformatic pipelines and especially the interpretation of data to confirm a definitive diagnosis differ between different GDCs. PIDs are heterogeneous and not exclusively monogenic disorders. Environmental and genetic risk factors play a role in phenotypic presentation. Furthermore, variations in specific genes that are important for immune defense only lead to disease when the patient is exposed to a specific pathogen. To ensure high-quality patient care, uniformity and concordance in variant interpretation among GDCs are required.
External quality assessments (EQA) have become an important aspect of laboratory medicine, and have aided the use of molecular diagnostics [9, 10]. An EQA usually consists of the distribution of the same patient samples to different laboratories for analysis and reporting, but some EQA schemes ask for a retrospective assessment. During retrospective assessments, known diagnostic cases are shared with laboratories.
No EQA studies have been performed within the PID field so far. A pilot EQA for severe combined immune deficiency (SCID) is planned by EMQN in 2020 [11]. Therefore, a quality assessment was performed between the four GDCs that perform PID diagnostics in the Netherlands. We aimed to compare variant interpretation outcomes between the different centers based on genetic tertiary data analysis.
Materials and methods
Participating centers and samples
Four Dutch GDC participated in this study: Radboud University Medical Center (Nijmegen (Department of Human Genetics, Division of Genome Diagnostics)), University Medical Center Utrecht (Utrecht, (Department of Genetics, Division Laboratories, Pharmacy and Biomedical Genetics)), Erasmus University Medical Center (Rotterdam (Department of Clinical Genetics)), and University Medical Center Groningen (Groningen (Department of Genetics)). All clinical laboratories are certified and accredited by ISO15189.
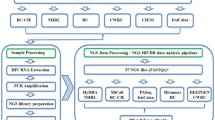
The process of sample distribution is illustrated in Fig. 1. Each GDC provided data for EQA from two DNA samples from real patients that underwent whole-genome sequencing (WGS) or whole-exome sequencing (WES) as part of evaluation for possible primary immunodeficiency. Laboratories provided one sample with a confirmed genetic diagnosis and one sample with an unknown genetic diagnosis. A genetic diagnosis was considered confirmed when one heterozygous pathogenic (P) or likely pathogenic (LP) classified variant was identified in case of a disease with autosomal dominant inheritance, or when a hemizygous variant was identified in case of X-linked (recessive) inheritance. In case of disease with a recessive inheritance, a genetic diagnosis was considered confirmed if one homozygous or two compound heterozygous P or LP classified variants were identified. This resulted in a total of eight samples (n = 4 confirmed genetic diagnoses and n = 4 unknown diagnoses). The unannotated VCF was shared for variant interpretation by clinical laboratory geneticists from each participating institution. Thus, every center analyzed six external samples. No detailed clinical data were shared, except for knowledge of an existing PID phenotype, as often is the case in practice. Metadata that could lead to identification of the patient was removed before sending the files.

Each site sent one sample with confirmed genetic diagnosis (represented as a) and one sample with unknown genetic diagnosis (represented as b) to three other sites, and received six samples from the other sites.
Workup per center
The characteristics of the NGS pipeline of every participating GDC are shown in Table 1. All samples underwent workup according to the analysis pipeline of their own center at the time the samples were initially processed in each center. The choice for a specific sequencing approach depended on availability in each institution. All centers performed either WES or WGS.
Following the objective of this study—to analyze uniformity in data interpretation of variants in NGS-based PID diagnostics based on VCF files—we did not compare performance of sequencing platforms or bioinformatic pipelines based on technical parameters. The results focused on the classification and reporting of variants and the uniformity thereof among different GDCs in the Netherlands. The description of NGS pipeline is provided for informative purposes and illustrated in Fig. 2.

The boxes left from the horizontal line are related to the presequencing, sequencing, and bioinformatics processes, and are not part of this study. These processes have been performed prior to the study. The boxes right from the horizontal line are related to variant interpretation processes and have been performed during this study.
Gene panel
In the Netherlands, a uniform gene panel for PIDs is used nationwide. The panel is updated by consensus meetings, primarily based on annual updates from the International Union of Immunological Societies [12]. The gene panel used in this study consisted of 389 genes (version 5, April 10th 2019). Only the genes in this panel were analyzed (Supplementary Table S1).
Variant interpretation and classification
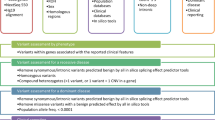
The participating clinical laboratory geneticists were asked to analyze the six unannotated VCF files they received from the other institutions using their own analysis pipeline and standard workflow, and to report all variants that would be reported to the medical specialist within the clinical setting. The centers were autonomous in the application of a specific strategy for providing an NGS-based diagnosis. Variant interpretation occurred according to the classification scheme of the American College of Medical Genetics, i.e., P, LP, variant of unknown significance (VUS), likely benign (LB), and benign (B) [6]. Only variants classified as P, LP, and—in some cases—VUS were reported, depending on autosomal dominance and relevance regarding phenotype. Per protocol, B and LB variants are never reported.
After interpretation, the results were compared and described. Specifically, variants that would be reported to the clinician or would need further investigation were compared between the centers, and discussed in a teleconference.
Survey of experiences of clinical laboratory geneticists
In order to evaluate the experiences of clinical laboratory geneticists during the EQA, a questionnaire was sent to all participants in the study (Supplementary Data). With the results of the study, we hoped to improve future EQA. The survey consisted of questions regarding the procedure of data entry and handling within the center, the analysis of a VCF file to create a report, the interpretation of data from other GDCs, and the additional information as was provided before analysis. Also, clinical laboratory geneticists were asked for suggestions to further improve future EQA.
Results
Variant interpretation and classification
Variants evaluated and classified by clinical laboratory geneticists that would be reported to the medical specialist are displayed in Table 2, accompanied by the associated phenotypes and pattern of inheritance according to the database Online Mendelian Inheritance in Man [13].
Despite the variety in approach and analyses, variants that would be included in diagnostic reports sent to the medical specialist were consistent over the four centers, except for eight variants. As shown in Table 2, the eight variants were located in the following genes: RFXANK, MEFV (CNV), IFIH1, NFKB2, GINS1, ADAR, LTBP3, and PLEKHM1.
All variants that were classified (L)P within its originating center were also classified as such by other centers, except for two variants. The PSTPIP1 nonsense variant p.(Gln219*) from site 1 was classified as VUS by all sites, except for a LP classification by site 2. Site 1 initially classified the PSTPIP1 variant p.(Gln219*) from their own VCF file as P because it concerned a truncating variant. Because all pathogenic variants in PSTPIP1 so far reported are activating missense variants, the final classification was changed to VUS. A BTK nonsense variant p.(Arg525*) from site 3 was classified P by all sites, except for a LP classification by site 2 because there was not enough evidence to classify it as P. Other differences in (L)P classifications occurred in a RFXANK variant p.(Ala63Thr) and a GINS1 variant, both reported by site 2 in the samples from sites 1 and 4, respectively. These variants were not reported and classified by the originating center.
Survey
The results of the survey among clinical laboratory geneticists revealed multiple suggestions for future EQA. All stated that the absence of BAM files and clinical information complicated the evaluation and interpretation of variants for multiple reasons. First, the absence of BAM files hampered the adequate assessment and exclusion of artifacts. Second, clinical information and phenotype of patients are essential to assess the relevance of specific variants. Without clinical information, variants might falsely be labeled irrelevant. For example, when a heterozygous variant is found in a gene for a recessive disease that would fit well with the phenotype of the patient. In that case, additional genetic testing should be advised to search for a missed second variant (e.g., a deletion) in that gene.
Other suggestions consisted of the standardization of the bioinformatic pipeline across centers to facilitate the exchange of data.
Discussion
We performed an EQA to assess uniformity of interpretation and reporting of variants in NGS-based PID diagnostics among Dutch GDC. To our knowledge, our study is the first EQA for NGS-based PID diagnostics, and provides an initial insight of the quality of genetic diagnostics in this field.
The variants that led to a clinical diagnosis and that would be included in a diagnostic report were largely consistent over the four centers; the majority of the variants classified as P or LP were identified and described correctly. In order to efficiently share variants, it is necessary to communicate the original genomic variant description next to the annotated description. The participating Dutch laboratories are used to share their variant data in this way as described by Fokkema et al. [14]. Discrepancies related to reporting and classifications between centers can be attributed to differences in analysis and filtering of variants after the sequencing process. Further synchronization of filter pipelines may be important, but organization thereof was beyond the scope of our study.
The results highlight the uniformity in data interpretation of variants in NGS-based PID diagnostics within the Netherlands, even without prior agreements on data interpretation. This might be attributed to the harmonization among Dutch clinical laboratory geneticists for the complete PID gene panel within PID diagnostics. During national meetings and conference calls, all participating clinical laboratory geneticists discuss, together with immunologists and clinical geneticists, specific inconclusive cases, and the reporting of specific variants. These regular meetings between the specialists have also led to a consensus and uniform PID gene panel that is discussed and agreed upon at least once a year, which is unique in Europe. All clinical laboratory geneticists work according to guidelines for NGS-based diagnostics [2]. The fact that participating clinical laboratory geneticists work according to a professional standard in a ISO15189-certified lab, contributes to high-quality patient care and uniformity in PID genetic diagnostics.
The nonsense variant in PSTPIP1 was classified as likely pathogenic by one site and classified as VUS by the three other sites. Moreover, the PSTPIP1 variant p.(Gln219*) was initially even classified as pathogenic by site 1. This shows that multidisciplinary discussions and data sharing are necessary for genetic testing in the NGS era to achieve correct and harmonized classifications among different laboratories. When it comes to variants classified as VUS, differences in reporting the variant to the medical specialist occurred, which seems to be dependent on the available clinical and immunological information of the patient. The possibility to perform pedigree analysis and/or immunological follow-up is an important next step for validating the biological meaning of genetic findings, and is highly relevant to understanding the disease manifestations in a given patient.
By using only VCF files for data analysis, it is difficult to discriminate between artifacts and real variants. Therefore, accurate BAM-file assessment or Sanger confirmation is essential to prevent misinterpretation and identify relevant variants with a low quality (true or false variant).
Also, a study of Gargis et al. [4] suggests that an important question during tertiary data analysis consists of whether a variant might be disease-causing, and to which extent the health outcome is relevant for the patient’s clinical presentation. However, specific information regarding patient phenotype was not made available during this EQA. In this study, clinical laboratory geneticists were unable to relate the genotype to the phenotype and immunological data, which complicated the decision whether a VUS was relevant to report. Especially in NGS-based diagnostics, a clear description of clinical phenotype and immunological test results is important due to several disease-specific factors.
First, many PIDs are not solely monogenic, meaning that environmental factors might influence the disease severity. Also, multiple genetic risk factors, known and unknown, may play a role in phenotypic presentation. Furthermore, variations in specific genes that are important for immune defense are not disease-causing but might—for instance—cause a higher susceptibility for specific infections. This shows that the variation itself is not disease-causing but might be when the patient is exposed to the specific pathogen. This complicates data interpretation, especially when essential clinical information about the patient is not present. An example of this situation occurred in the MBL gene [15]. This gene possesses several common SNPs that affect functioning of the mannose-binding lectin 2, and variants of the MBL gene have shown to be associated with an increased susceptibility of infections. This implies that solely the variations of MBL are not disease-causing in itself. This, again, highlights the importance and necessity of clinical information of patients within PID diagnostics.
Second, many primary immune deficiencies show variable penetrance of symptoms that can range from milder forms to severe phenotypes of disease. In this spectrum, modifying factors play a major role, resulting in different expression levels of genes in the affected pathways. For instance, adenosine deaminase-1 (ADA1) deficiency affects lymphocyte development and function [16]. The phenotypic spectrum ranges from occurrence of SCID, which is usually diagnosed in children aged 6–12 months, to partial ADA deficiency with mild and benign phenotypes.
Within our study, a PSTPIP1 variant p.(Gln219*) was reported, but classified differently by different sites. Variants in PSTPIP1, or CD2BP1, are associated with the autosomal dominantly inherited PAPA (pyogenic sterile arthritis, pyoderma gangrenosum, and acne) syndrome [17]. Disease-causing variants of this gene might jeopardize the mechanism responsible for maintenance of a proper inflammatory response. However, it is unknown if loss-of-function variants within PSTPIP1 gene are causing a phenotype. Adequate clinical patient information can support a molecular diagnosis. The role of non-Mendelian inheritance patterns might be of great importance for these cases [18].
Last, many of the symptoms that PID patients present with (e.g., fever and infections) are highly frequent in the general population. If no complete clinical phenotype is described in genetic testing request, a link from a possible disease-causing genetic variant to the clinical phenotype of the patient might not be recognized. For instance, TWEAK deficiency caused by a genetic defect in TNFSF12, might cause common variable immunodeficiency (CVID) accompanied by nonspecific clinical manifestations such as pneumonia and warts [17, 19]. However, a recent study shows that warts are the most common (41.3%) among common skin diseases in Europe [20]. This complicates the adequate interpretation of gene variants without proper clinical information.
Other limitations of our study include the essential aspects of EQAs in themselves. When performing EQAs, Hastings and Howell [21] emphasize the importance of predefined criteria. The participating centers did work according to existing guidelines, and used a PID gene panel based on consensus meetings and literature [2, 12]. However, a limitation of this study might be that predefined criteria for analysis were not present prior to the EQA, and all centers worked according to their own strategy. Furthermore, EQAs usually simulate the existing diagnostic processes before interpretation takes place [21]. This implies that patient materials and clinical information are distributed among the participating centers. Because we only distributed unannotated VCF files within this study and a minimum of clinical information, the interpretation of the variants might have been hampered.
In conclusion, tertiary data analysis was largely consistent among Dutch GDC, and can be used to assess uniformity in data interpretation. However, the technical possibility to discriminate between artifacts and real variants, and the availability of sufficient clinical and immunological information, is necessary for proper interpretation of genetic data. International sharing and discussing of variant data, in addition to an international EQA for PID, could further harmonize variant interpretation and thus improve the quality of diagnosis and care for PID patients.
References
Di Resta C, Galbiati S, Carrera P, Ferrari M. Next-generation sequencing approach for the diagnosis of human diseases: open challenges and new opportunities. Electron J Int Fed Clin Chem Lab Med. 2018;29:4–14.
Weiss MM, Van der Zwaag B, Jongbloed JDH, Vogel MJ, Brüggenwirth HT, Lekanne Deprez RH, et al. Best practice guidelines for the use of next-generation sequencing applications in genome diagnostics: a national collaborative study of dutch genome diagnostic laboratories. Hum Mutat. 2013;34:1313–21.
Heimall JR, Hagin D, Hajjar J, Henrickson SE, Hernandez-Trujillo HS, Tan Y, et al. Use of genetic testing for primary immunodeficiency patients. J Clin Immunol. 2018;38:320–9.
Gargis AS, Kalman L, Bick DP, Da Silva C, Dimmock DP, Funke BH, et al. Good laboratory practice for clinical next-generation sequencing informatics pipelines. Nat Biotechnol. 2015;33:689–93.
Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, et al. The variant call format and VCFtools. Bioinformatics 2011;27:2156–8.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24.
Arts P, Simons A, Alzahrani MS, Yilmaz E, AlIdrissi E, van Aerde KJ et al. Exome sequencing in routine diagnostics: a generic test for 254 patients with primary immunodeficiencies. Genome Med. 2019. https://doi.org/10.1186/s13073-019-0649-3.
Haer-Wigman L, van der Schoot V, Feenstra I, Vulto-Van Silfhout AT, Gilissen C, Brunner HG et al. 1 in 38 individuals at risk of a dominant medically actionable disease. Eur J Hum Genet. 2019. https://doi.org/10.1038/s41431-018-0284-2.
Davies KD, Farooqi MS, Gruidl M, Hill CE, Woolworth-Hirschhorn J, Jones H, et al. Multi-institutional FASTQ file exchange as a means of proficiency testing for next-generation sequencing bioinformatics and variant interpretation. J Mol Diagn. 2016;18:572–9.
Rossen JWA, Friedrich AW, Moran-Gilad J. Practical issues in implementing whole-genome-sequencing in routine diagnostic microbiology. Clin Microbiol Infect. 2018;24:355–60. https://doi.org/10.1016/j.cmi.2017.11.001.
ERN RITA. NEW! Severe Combined Immunodeficiency (SCID) EQA Scheme 2020. http://rita.ern-net.eu/2019/04/25/new-severe-combined-immunodeficiency-scid-eqa-scheme-2020/. Published 2019. Accessed October 29, 2019.
Bousfiha A, Jeddane L, Picard C, Ailal F, Gaspar HB, Al-Herz W, et al. The 2017 IUIS Phenotypic Classification for Primary Immunodeficiencies. J Clin Immunol. 2018;38:129–43.
McKusick VA. Mendelian Inheritance in Man and its online version, OMIM. Am J Hum Genet. 2007;80:588–604.
Fokkema IFAC, van der Velde KJ, Slofstra MK, Ruivenkamp CAL, Vogel MJ, Pfundt R et al. Dutch genome diagnostic laboratories accelerated and improved variant interpretation and increased accuracy by sharing data. Hum Mutat. 2019. https://doi.org/10.1002/humu.23896.
Garred P, Larsen F, Seyfarth J, Fujita R, Madsen HO. Mannose-binding lectin and its genetic variants. Genes Immun. 2006. https://doi.org/10.1038/sj.gene.6364283.
Hershfield M. Adenosine Deaminase Deficiency. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A E, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle. https://www.ncbi.nlm.nih.gov/books/.
Picard C, Bobby Gaspar H, Al-Herz W, Bousfiha A, Casanova JL, Chatila T, et al. International Union of Immunological Societies: 2017 Primary Immunodeficiency Diseases Committee Report on Inborn Errors of Immunity. J Clin Immunol. 2018;38:96–128.
Schejbel L, Garred P. Primary immunodeficiency: complex genetic disorders? Clin Chem. 2007;53:159–60.
Bogaert DJA, Dullaers M, Lambrecht BN, Vermaelen KY, De Baere E, Haerynck F. Genes associated with common variable immunodeficiency: one diagnosis to rule them all? J Med Genet. 2016. https://doi.org/10.1136/jmedgenet-2015-103690.
Svensson A, Ofenloch RF, Bruze M, Naldi L, Cazzaniga S, Elsner P et al. Prevalence of skin disease in a population-based sample of adults from five European countries. Br J Dermatol. 2018. https://doi.org/10.1111/bjd.16248.
Hastings RJ, Howell RT. The importance and value of EQA for diagnostic genetic laboratories. J Community Genet. 2010;1:11–7.
Acknowledgements
This publication and the underlying study have been made possible partly on the basis of the data that Hartwig Medical Foundation and Central Genome Analysis Laboratory—UMC St. Radboud have generated for the study. We thank our colleague Walter de Valk from the Erasmus Medical Center and Genomics Coordination Center—UMC Groningen for his support regarding bioinformatics.
Funding
The external quality assessment (EQA) is part of the Genetics First: next-generation sequencing for cost-effective PID diagnostics project. This project is funded by ZonMw (grant No. 846002001).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Elsink, K., Huibers, M.M.H., Hollink, I.H.I.M. et al. National external quality assessment for next-generation sequencing-based diagnostics of primary immunodeficiencies. Eur J Hum Genet 29, 20–28 (2021). https://doi.org/10.1038/s41431-020-0702-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-020-0702-0
This article is cited by
-
Utility of targeted next generation sequencing for inborn errors of immunity at a tertiary care centre in North India
Scientific Reports (2022)
-
Next-Generation Sequencing in the Field of Primary Immunodeficiencies: Current Yield, Challenges, and Future Perspectives
Clinical Reviews in Allergy & Immunology (2021)
