
Abstract
Defects in optic fissure closure can lead to congenital ocular coloboma. This ocular malformation, often associated with microphthalmia, is described in various clinical forms with different inheritance patterns and genetic heterogeneity. In recent times, the identification of an increased number of genes involved in numerous cellular functions has led to a better understanding in optic fissure closure mechanisms. Nevertheless, most of these genes are also involved in wider eye growth defects such as micro-anophthalmia, questioning the mechanisms controlling both extension and severity of optic fissure closure defects. However, some genes, such as FZD5, have only been so far identified in isolated coloboma. Thus, to estimate the frequency of implication of different ocular genes, we screened a cohort of 50 patients affected by ocular coloboma by using targeted sequencing of 119 genes involved in ocular development. This analysis revealed seven heterozygous (likely) pathogenic variants in RARB, MAB21L2, RBP4, TFAP2A, and FZD5. Surprisingly, three out of the seven variants detected herein were novel disease-causing variants in FZD5 identified in three unrelated families with dominant inheritance. Although molecular diagnosis rate remains relatively low in patients with ocular coloboma (14% (7/50) in this work), these results, however, highlight the importance of genetic screening, especially of FZD5, in such patients. Indeed, in our series, FZD5 variants represent half of the genetic causes, constituting 6% (3/50) of the patients who benefited from a molecular diagnosis. Our findings support the involvement of FZD5 in ocular coloboma and provide clues for screening this gene during current diagnostic procedures.
Similar content being viewed by others


Introduction
The human eye development is a complex and well-conserved vertebrate embryological process. Broadly, it becomes visible during the 4th week of development from an initial interaction between the optic vesicle (derived from neuroepithelium) that will evolve in a double-layered optic cup, and the lens placode (derived from the surface ectoderm), which will in the meantime invaginate to form the lens vesicle [1, 2]. During this asymmetric process, a transient opening on the ventral face of the optic cup appears and allows the vascular and nervous systems to get through; it will finally close around the 7th week of embryonic life [3, 4]. This morphogenetic process occurs according to three different axes (proximal–distal, dorsal–ventral and nasal–temporal), involving many signalization pathways and a complex and intricated genetic regulation [1, 5].
Congenital ocular anomalies result from a defect in the molecular and cellular mechanisms of eye formation. For example, those affecting the entire ocular globe can lead to the severe spectrum of anophthalmia and microphthalmia (absence and reduced growth of the ocular globe, respectively) with a prevalence of 1–3 per 10,000 births [6]. Ocular coloboma consists in a defect of optic fissure closure [1,2,3]. The closure process requires first a correct alignment of the fissure margins, ensured by a correct patterning along the three axes of the optic cup [1]. Then, the fusion phenomenon can start at the midpoint to progress both distally and proximally, thanks to multiple processes: changes in cell morphology at the optic fissure, basement membrane dissolution and cell adhesion, apoptosis phenomena, all these interacting with periocular mesenchyme cells [1, 3, 4]. Given the optic fissure location, coloboma is de facto located in the infero-nasal quadrant, potentially affecting any structure from the iris to the optic nerve [3, 7]. As a consequence, the phenotype may vary from normal vision to blindness forms [3]. In addition, ocular coloboma can be unilateral or bilateral, isolated or associated with other ocular malformations such as microphthalmia and anophthalmia (M/A) or with extraocular features in syndromic forms, such as CHARGE syndrome [MIM#214800] or Baraitser-Winter syndrome (BRWS) [MIM#243310] as well in several chromosomal aberrations [3, 7]. Moreover, inheritance varies from sporadic to familial cases with autosomal dominant, autosomal recessive or X-linked inheritance patterns reported. Worldwide prevalence ranges from 2 to 14 per 100,000 births causing 20% of childhood visual impairment [8]. Causes are multiple, from primary (congenital) to secondary, such as consequence of surgery, or environmental forms. Distinction is drawn between ocular coloboma and “atypical” coloboma resulting from iris defects whose genetic cause is different (mainly variants in PITX2, FOXC1 and PAX6) [2, 3]. The large majority of the 30 genes described as implicated in ocular coloboma are syndromic and non-syndromic M/A genes [1, 6, 9]. This clinical overlap is often entitled with the term “MAC” spectrum (M/A and Coloboma).
Interestingly, two genes have been so far identified in patients with isolated ocular coloboma: FZD5 [MIM*601723] and a recurrent variant in ACTG1 [MIM*102560] [10, 11]. FZD5 (frizzled class receptor 5) has been involved in a single dominant form of coloboma, after whole-exome sequencing (WES) was performed in a large family, identifying an ultra-rare disease-causing variant in 13 individuals with either non-syndromic bilateral chorioretinal or iris coloboma (n = 10), unilateral optic disc (OD) abnormality (n = 1), or without any ocular phenotype (n = 2), considered as non-penetrant allele [10]. FZD5 encodes for the FZD5 protein, which is a Wnt receptor that mediates canonical and non-canonical Wnt-signaling pathways [12]. The frameshift variant c.656delCinsAG [p.(Ala219Glufs*49)] detected in FZD5 was shown to produce a truncated protein lacking the transmembrane domain (TMD), but with a conserved extracellular cysteine-rich Wnt-binding domain (CRD) [10]. The authors first showed that a disruption in the Wnt-FZD5 signaling pathway leads to a defect in optic fissure closure in humans, as already demonstrated in animal models [10, 13, 14]. Regarding ACTG1, it was formerly identified as the BRWS responsible gene [15]. Interestingly, a de novo ACTG1 variant c.209C > T [p.(Pro70Leu)] was detected in two unrelated patients with bilateral iris and chorioretinal coloboma with no extraocular BRWS feature [11].
Thus, to identify the underlying genetic cause in a cohort of 50 patients affected with coloboma of unknown etiology, we performed a targeted resequencing approach using a 119 ocular developmental genes panel.
Materials and methods
Cohort description
This study was designed in compliance with the tenets of the Helsinki Declaration. Informed consent was obtained from all individuals included in this study.
A cohort of 50 patients with a clinical diagnosis of unilateral or bilateral coloboma in a familial (n = 11) or sporadic inheritance pattern (n = 39) was analyzed on a targeted NGS panel of 119 genes involved in various ocular defects. This targeted resequencing panel contains different categories of ocular developmental genes (Suppl.data1). In particular, it includes all the coloboma-associated genes described to date, as well as all the genes involved in overlapping phenotypes such as micro-anophthalmia (MAC spectrum) or anterior segment dysgeneses responsible of iris defects that can mimic iris coloboma. Indeed, among these 50 patients, 11 were displaying bilateral coloboma associated with unilateral (4/11) or bilateral (7/11) microphthalmia and 11 patients were presenting with a coloboma affecting only the iris. Extraocular features were observed in 11 out the 50 patients for which no association was diagnosed as part of a known syndrome. Clinical and molecular data are summarized in Table 1. Additional array CGH was performed in patients with associated features such as intellectual disability and/or abnormalities (Table 1).
Targeted NGS panel analysis
The 50 patients’ cohort was screened by means of a customized NGS panel including 119 ocular developmental genes (Suppl.data1). DNA was extracted from blood using the MagnaPure system (Roche Applied Science, Germany). Capture probes were designed with SureDesign (Agilent, USA). A library of all coding exons and intron-exon boundaries was prepared using the SureSelect XT HS and XT Low Input Enzymatic Fragmentation Kit and the SureSelect XT HS Target Enrichment System for Illumina Paired-End Multiplexed Sequencing Library (Agilent, USA) following the manufacturer’s instructions. Further sequencing was performed on a NextSeq500 plateform (Illumina Inc., CA). Sequence alignment was performed with BWA 0.7.10, picard-tools-2.18.23, elprep4 (Indel realignment, base recalibration). Then, the variant calling was made with GATK-3.3 (HaplotypeCaller) and Varscan2.3.7. Annotations were made with SNPEff-4.3 with additional information from gnomAD, ClinVar, and dbSNP151.
All variants were confirmed by Sanger sequencing. Sequence variants were numbered with the Adenine of the ATG initiation codon considered as the first nucleotide. GenBank references were: FZD5: NM_003468.4, MAB21L2: NM_006439, PTCH1: NM_000264, RARB: NM_000965, RBP4: NM_006744, and TFAP2A: NM_003220.
All the novel variants described in this study have been submitted to ClinVar database.
Assessment of pathogenicity
Variants were classified according to the ACMG guidelines [16]. All “Pathogenic”, “likely pathogenic” and “of unknown significance” (VUS) variants were reported in this study. “Pathogenic” and “likely pathogenic” variants were considered as disease-causing variants when familial segregation of the variant was in accordance with the mode of inheritance. Of note, no FZD5 transcript analysis could be performed.
Results
This exhaustive genetic analysis revealed seven disease-causing variants, respectively in RARB, MAB21L2, TFAP2A, RBP4 and FZD5, as well as one variant of uncertain significance in PTCH1, in 8 out the 50 probands (Table 1).
Likely pathogenic variants in FZD5
Three different FZD5 likely pathogenic variants, never described before, were detected in three unrelated families with dominant inheritance. They were considered as disease-causing variants. Clinical and molecular data are detailed in Table 2.
Patient 1: sporadic bilateral non-syndromic coloboma
Patient 1 (ADN120143) was a 10-year-old girl, the first child of unrelated Algerian and Moroccan parents with no familial history of ocular malformation (apart from a right isolated convergent squint in the father, without coloboma). She was diagnosed with a right iris, choroidal and OD coloboma adjacent to the macula and a left choroidal and OD coloboma at 10 months of life during an ophthalmological examination for a right convergent squint. Neurocognitive development and growth parameters were unremarkable. She had no other malformation and her clinical examination showed no abnormality including the absence of distinctive craniofacial features. A heterozygous frameshift variation c.820del [p.(Leu274Cysfs*73)] was detected in FZD5 (Suppl.data2). This variation was never reported in the literature and was absent from gnomAD. Complete segregation pattern could not be established due to the unavailability of the DNA’s father. Following the ACMG guidelines [16], the variant is considered as likely pathogenic (PVS1, PM2). The ClinVar accession number for this variant is SCV000998820.
Patient 2: sporadic bilateral syndromic coloboma
Patient 2 (ADN140259) was a 43-year-old woman, the fourth out of six siblings, born from unrelated Algerian parents, herself mother of four children. Ocular familial history was unremarkable. She was diagnosed at birth with a right OD coloboma and a left iris and OD coloboma associated with a cortical cataract causing blindness. She suffers from bilateral congenital deafness with cochlear malformations (no further information available) and neurocognitive difficulties (trouble in learning, memorization, concentration). Cerebral MRI showed a postero-inferior and internal herniation of the left eyeball in contact with the optic nerve. Array CGH found no chromosomal imbalance. A heterozygous in-frame variant c.1081_1082insGAA [p.(His361delinsArgAsn)] was identified in FZD5. This variation was never reported in the literature and was absent from gnomAD. This in-frame codon insertion happens in the major functional frizzled domain of the protein (UniProt: Q13467). The loss of the highly conserved amino acid Histidine (Suppl.data2) with the insertion of two novel amino acids Arginine and Asparagine is predicted to be damaging by in silico analyses (Human Splicing Finder, GeneSplicer, ESEfinder). Due to the unavailability of parental DNA, segregation pattern could not be established. According to the ACMG guidelines [16], the variant is considered as likely pathogenic (PM1, PM2, PM4 and PP3). The ClinVar accession number for this variant is SCV000998821.
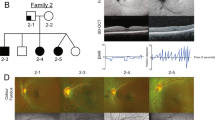
Patients 3 and 4: familial bilateral isolated coloboma
Patient 3 (SG170145), a 26-year-old woman and her mother (patient 4) are the only two symptomatic members in their family of French origin. The daughter (patient 3) has a bilateral iris and chorioretinal coloboma affecting the macula and the optic nerve, with secondary congenital nystagmus and high myopia (−11 diopters in the right eye, −4.75 diopters in the left eye). Her mother has a bilateral OD coloboma. In both affected members of this family, a heterozygous in-frame deletion of 22 amino acids c.1181_1246del [p.(Asn394_Gly415del)] was identified, reaching the Frizzled domain of FZD5. The core functional domain is deprived of 22 highly conserved amino acids (Suppl.data2) and was predicted to be damaging by in silico analyses (Human Splicing Finder, Gene splicer, MaxEnt, EX-SKIP). This variant was never reported in the literature and is absent from gnomAD. According to the ACMG guidelines [16], the variant is considered as likely pathogenic (PM1, PM2, PM4 and PP3). The ClinVar accession number for this variant is SCV000998822.
(Likely) pathogenic variants in RARB, MAB21L2, RBP4 and TFAP2A
A de novo RARB missense variant (NM_000965: c.872A > T, [p.(His291Leu)]) was identified in patient 16 (ADN150083), a 6-year-old boy with bilateral iris coloboma and diaphragmatic hernia. De novo missense variants in RARB are known to be deleterious and involved in RARB-related PDAC syndrome [17]. This variant was predicted to be damaging by in silico analyses (SIFT, PolyPhen-2, MutationTaster). It was never reported in the literature and is absent from gnomAD. According to the ACMG guidelines [16], the variant is considered as likely pathogenic (PS2, PM1, PM2 and PP3). The ClinVar accession number for this variant is SCV000803842.
A novel heterozygous null variant in MAB21L2 (NM_006439: c.1A > C; p.[Met1?]) was then identified in patient 40 (LGN120039), a 30-year-old women with bilateral colobomatous microphthalmia, intellectual disability, autistic features, craniofacial dysmorphy with scaphocephaly, but without rhizomelic skeletal dysplasia as described in the initial report [18]. Of note, array CGH was normal. Her father was also heterozygous for the c.1A > C, but affected only by isolated bilateral colobomatous microphthalmia. This null variant affecting the initiation codon was never reported in the literature and is absent from gnomAD. According to the ACMG guidelines [16], the variant is considered as likely pathogenic (PVS1 and PM2). The ClinVar accession number for this variant is SCV001364581.
A heterozygous RBP4 variant (NM_006744: c.569-1G > A, p.?) was identified in patient 27 (SG102327), a 75-year-old men with isolated and sporadic bilateral retinal coloboma. This null variant was never reported in the literature and is absent from gnomAD. Due to the unavailability of parental DNA, segregation pattern could not be established. According to the ACMG guidelines [16], the variant is considered as likely pathogenic (PVS1 and PM2). The ClinVar accession number for this variant is SCV001364582.
Finally, an heterozygous TFAP2A variant (NM_003220: c.1037_1038del, [p.(Lys346fs*84)]) was identified in patient 50 (SG130194), a 7-year-old girl presenting with bilateral OD coloboma and unilateral microphthalmia as her mother and maternal grandfather. This null variant, transmitted by the symptomatic mother, is absent from gnomAD and was once reported in the Clinvar database as pathogenic (SCV000747548.1). According to the ACMG guidelines [16], the variant is considered as pathogenic (PVS1 and PM2). The ClinVar accession number for this variant is SCV001364583.
Variant of uncertain significance in PTCH1
A heterozygous PTCH1 variant (NM_000264: c.2500T > G, p.Leu834Val) was identified in patient 23 (SG080744), a 15-year-old girl presenting with isolated and sporadic bilateral ocular coloboma (right iris coloboma and left macular and OD coloboma). It was predicted to be damaging by in silico analyses (SIFT, PolyPhen-2, MutationTaster). This variant was never reported in the literature and is absent from gnomAD. Due to the unavailability of parental DNA, segregation pattern could not be established. According to the ACMG guidelines [16], the variant is considered as being of uncertain significance (PM1, PM2, and PP3). The ClinVar accession number for this variant is SCV001364584.
Discussion
Herein, we tested 50 patients with a phenotype of ocular coloboma on a targeted NGS panel of 119 ocular developmental genes. The ocular coloboma phenotype was either “purely isolated” (n = 31) or associated with other eye defects (mainly microphthalmia) (n = 8), with systemic anomalies (n = 8) or both (n = 3). Twenty-two percent (11/50) were familial forms.
Four different (likely) pathogenic variants were identified, respectively, in RARB, MAB21L2, TFAP2A, and RBP4, genes repeatedly implicated in MAC phenotypes [6], as well as a variant of uncertain significance in PTCH1. Two interesting observations have to be made about the RARB and MAB21L2 variants identified in this work.
Firstly, de novo missense variants in RARB are known to be responsible for the PDAC syndrome through a gain-of-function mechanism [6, 17]. However, although our patient’s genotype is classic, his phenotype is less so. Indeed, the absence of eye growth defects in patient 16 (ADN150083) is an infrequent feature that has only been reported once in an individual with a RARB variant (c.431G > A, p.Arg144Gln) inherited from his mother whom had herself a colobomatous microphthalmia [19].
Then, concerning MAB21L2, the genotype is as unusual as the phenotype. Here, we reported the first heterozygous MAB21L2 variant affecting the initiation codon (c.1A > C) in a gene where almost only missense variants have been described [6]. Solely one nonsense variant (c.840C > G, p.Tyr280Ter) was described by Patel et al. in MAB21L2 [20] in a patient with isolated colobomatous microphthalmia. Thus, this report of a second null variant in MAB21L2 is in correlation with the idea that, unlike missense variants that are located in a mutational hotspot which may have a crucial role both in eye and skeletal development [6], null variants in MAB21L2 could exert their effects only on ocular development.
Besides the identification of variants in RARB, MAB21L2, TFAP2A, and RBP4, as well as PTCH1, three novel disease-causing FZD5 variants were identified in this study.
Of note, the recurrent ACTG1 variant was not detected in our cohort. This is in full accordance with the low frequency observed from the initial description (two patients in around 400 tested) [11] and a recent study testing 66 familial cases of coloboma [19].
This work allows describing three novel heterozygous FZD5 likely pathogenic variants considered as disease-causing: one frameshift c.820del [p.(Leu274Cysfs*73)], one in-frame insertion c.1081_1082insGAA [p.(His361delinsArgAsn)] and one in-frame deletion c.1181_1246del [p.(Asn394_Gly415del)] in patients with either isolated coloboma or associated with extraocular features. Surprisingly, our 6% variant detection rate (3/50) is far superior to the previously described, with only one variant identified in 552 patients (172 + 380 patients from two cohorts) with optic fissure defects screened for FZD5 variants [10].
FZD5 is a single coding exon gene located in the 2q33.3 region, which encodes for the FZD5 protein, a transmembrane receptor for Wnt ligand, composed of an extracellular CRD domain and a transmembrane Frizzled domain (NCBI database). The variant c.656delCinsAG [p.(Ala219Glufs*49)] previously described and located in the linker region (Fig.1) results in the production of a truncated protein of 267 amino acids lacking the TMD, with a dominant-negative effect demonstrated by functional analyses [10]. Interestingly, all three novel variants identified herein consist also in small insertions and deletions (Indels) and each of them is affecting one out of the seven TMDs of the frizzled domain (Fig. 1). Because of the closely location of the previously described variant, a dominant-negative effect can be as well a plausible mechanism for the c.820del variant [p.(Leu274Cysfs*73)] identified in patient 1, with, possibly, a production of a truncated protein of 346 amino acids. Effects of the two others, the c.1081_1082insGAA [p.(His361delinsArgAsn)] and the c.1181_1246del [p.(Asn394_Gly415del)], impacting the fourth and the fifth TMD, respectively, have to be determined more precisely, given that a large part of the TMD would be, this time, conserved. At this point, two other mechanisms could also be considered for all these variants: either a loss-of-function effect or nay, a gain-of-function effect, as already suggested in the work of Liu et al. in 2016 [10]. The mechanisms by which the FZD5 variants lead to coloboma remain elusive. As guidance, no clear genotype–phenotype correlation emerged with the FZD4 paralogue gene, whose variants lead to autosomal dominant familial exudative vitreoretinopathy [21]. Interestingly, FZD4 has a highly specific ligand, Norrin, which signaling is lost when swapping FZD4 linker region (located between the CRD and the Frizzled domain) with the FZD5 one, suggesting the importance of this region in ligand-specific cellular response [22]. This linker region could also have an importance in FZD5 function.

In bold, the novel variants reported here.
Moreover, the gnomAD database contains seven rare truncating variants (with an allele frequency ranging from 1/31,384 to 1/248,778) in the FZD5 gene, located in the linker region and the Frizzled domain, all respecting the CRD. The occurrence of such variants in an “assumed” asymptomatic population could be explained by either (i) an incomplete penetrance of these variants as illustrated by the original family reported by Liu et al. [10], or (ii) a mild ocular phenotype in those patients (visual impairment ranges from asymptomatic to complete loss of vision, depending on the coloboma size and location).
Afterwards, this work allows expanding the phenotypic spectrum of FZD5 variants, in particular showing the intra/inter-familial variability but also the intra-individual variability such as in patients 1 and 2, who display a different coloboma pattern depending on the eye side. Although the eye phenotype is not detailed for each mutated member of the family described by Liu et al. [10], relevant points regarding the eye phenotype of our mutated patients can be highlighted: (i) the absence of a coloboma affecting solely the iris but rather (ii) the presence in all of a coloboma affecting the optic nerve (like in most of the PAX2 mutated patients [1]), (iii) the absence of associated M/A and, finally, (iv) the absence of retinal involvement in view of the functional work done by Liu et al. showing the retinal developmental defect when the Wnt-FZD5 signaling pathway is altered [10]. Moreover, we can notice that patient 2, carrying the c.1081_1082insGAA [p.(His361delinsArgAsn)], displayed a coloboma with extraocular features (bilateral congenital deafness and neurocognitive difficulties). Of note, other defects in Wnt-signaling components are involved in syndromes associating ocular abnormalities, deafness and intellectual deficiency, like in the recessive X-linked Norrie disease [MIM#310600]. Besides, supported by a mice FZD4−/− model, Norrin–FZD4 signaling system has been shown to play a central role in vascular development in the eye and ear, allowing us to assume a link between our patient phenotype and a defect in the Frizzled-protein Wnt-signaling pathway [23]. Unless incidental, it is also interesting to note that FZD5 is, for the first time, involved in an associated form of coloboma, assuming the first description of a syndromic form. As no genotype–phenotype correlation seems to emerge (at least for the moment given the low number of reported variants), it is concordant with the high clinical variability observed with most of the genes involved in ocular growth and structural defects. Although occasional, other genes were already described either in syndromic and non-syndromic forms of coloboma, such as SOX2 or PAX6 [1, 6].
Thus, the FZD5 gene is responsible for the coloboma phenotype in 3 out of the 50 families and its implication therefore represents 6% of our cohort, which can be assumed to explain a relatively “high” proportion of cases. Indeed, most of the time, patients with non-syndromic coloboma (>70%) have no identified genetic cause and, despite a wide genetic heterogeneity, each gene explains only a small percentage of cases (less than 3% each) [11]. In our cohort, only 14% of patients (7/50) benefited from a molecular diagnosis. This is in correlation with a recent study that reported a diagnosis rate of 4.5% after screening 38 MAC genes in 66 families affected with syndromic and non-syndromic coloboma [19]. In other words, the chance for identifying a genetic underlying cause is weak in the presence of coloboma. This is well illustrated in the cohort of the 150 MAC patients described by Chassaing et al., in which a genetic etiology was proved only in 7% of the 56 colobomatous microphthalmia cases, against 17% of the 53 non-colobomatous microphthalmia cases and 54% of the 41 anophthalmia cases [9]. Nevertheless, we bring here important pieces of evidence for considering FZD5 as an important coloboma gene and therefore new insights for the existence of Mendelian genetic bases for the ocular coloboma phenotype.
Facing these latter results, we can also wonder whether microphthalmia, when associated and moderate, is not simply the consequence of a large coloboma. It is classically admitted that structural eye defects of the MAC spectrum represent a phenotypic continuum, and that eye growth defect is a mechanism for coloboma occurrence because fissure margins cannot appose properly. But other mechanisms could be possible, like failure of epithelial fusion. Thus, the coloboma phenotype could be considered as a full clinical entity and the associated microphthalmia as a consequence in the manner of a “sequence”. In addition, the relatively “high” frequency of FZD5 involvement in coloboma phenotype definitely contrasts with the absence of FZD5 variants identified to date in anophthalmia and non-colobomatous microphthalmia patients. M/A phenotypes are genetically explained in <50% of the patients [6]. Even novel sequencing technologies, such as WES studies, in cohorts of M/A patients, resulted in a detection rate similar to that obtained by the targeted sequencing of a panel of the main known genes [6].
Advances in genetics have to be made in the field of optic fissure closure, both to improve the diagnosis rate and genetic counseling delivered to the patients and their families. The detection rate by using screening of genes known to be involved in non-syndromic ocular developmental defects remains low with regard to the familial cases (22% in this study). Adding FZD5 is useful and cost-effective. Some new genes will probably be identified in the future, in association with the generalization of WES studies approaches. However, other molecular non-mendelian mechanisms, such as gene expression regulation or epigenetic processes, could exist and wait to be discovered by using whole genome or methylation studies. Although progress need to be done, FZD5 can from now be considered as a major gene of ocular coloboma and added in NGS panel dedicated to coloboma diagnostic procedures.
URLs
*ClinVar: https://www.ncbi.nlm.nih.gov/clinvar/
*ESEfinder: http://krainer01.cshl.edu/cgi-bin/tools/ESE3/esefinder.cgi?process=home
*ELprep: https://github.com/ExaScience/elprep
*GenBank: https://www.ncbi.nlm.nih.gov/genbank/
*GnomAD: http://gnomAD.broadinstitute.org
*Gene splicer: https://ccb.jhu.edu/software/genesplicer/
*Human Slicing Finder: http://www.umd.be/HSF
*NCBI Database: https://www.ncbi.nlm.nih.gov/
References
Patel A, Sowden JC. Genes and pathways in optic fissure closure. Semin Cell Dev Biol. 2019;91:55–65.
Graw J. Eye development. Curr Top Dev Biol. 2010;90:343–86.
Onwochei BC, Simon JW, Bateman JB, Couture KC, Mir E. Ocular colobomata. Sur Ophthalmol. 2000;45:175–94.
Gestri G, Bazin-Lopez N, Scholes C, Wilson SW. Cell behaviors during closure of the choroid fissure in the developing eye. Front Cell Neurosci. 2018;12:42.
Zagozewski JL, Zhang Q, Eisenstat DD. Genetic regulation of vertebrate eye development. Clin Genet. 2014;86:453–60.
Plaisancie J, Ceroni F, Holt R, Zazo Seco C, Calvas P, Chassaing N, et al. Genetics of anophthalmia and microphthalmia. Part 1: non-syndromic anophthalmia/microphthalmia. Hum Genet. 2019;138:799–830.
AS AL, Gregory-Evans CY, Gregory-Evans K. An update on the genetics of ocular coloboma. Hum Genet. 2019;138:865–80.
Shah SP, Taylor AE, Sowden JC, Ragge NK, Russell-Eggitt I, Rahi JS, et al. Anophthalmos, microphthalmos, and typical coloboma in the United Kingdom: a prospective study of incidence and risk. Invest Ophthalmol Vis Sci. 2011;52:558–64.
Chassaing N, Causse A, Vigouroux A, Delahaye A, Alessandri JL, Boespflug-Tanguy O, et al. Molecular findings and clinical data in a cohort of 150 patients with anophthalmia/microphthalmia. Clin Genet. 2014;86:326–34.
Liu C, Widen SA, Williamson KA, Ratnapriya R, Gerth-Kahlert C, Rainger J, et al. A secreted WNT-ligand-binding domain of FZD5 generated by a frameshift mutation causes autosomal dominant coloboma. Hum Mol Genet. 2016;25:1382–91.
Rainger J, Williamson KA, Soares DC, Truch J, Kurian D, Gillessen-Kaesbach G, et al. A recurrent de novo mutation in ACTG1 causes isolated ocular coloboma. Hum Mutat. 2017;38:942–6.
Fujimura N. WNT/beta-catenin signaling in vertebrate eye development. Front Cell Dev Biol. 2016;4:138.
Liu C, Nathans J. An essential role for frizzled 5 in mammalian ocular development. Development. 2008;135:3567–76.
Liu C, Bakeri H, Li T, Swaroop A. Regulation of retinal progenitor expansion by Frizzled receptors: implications for microphthalmia and retinal coloboma. Hum Mol Genet. 2012;21:1848–60.
Riviere JB, van Bon BW, Hoischen A, Kholmanskikh SS, O’Roak BJ, Gilissen C, et al. De novo mutations in the actin genes ACTB and ACTG1 cause Baraitser-Winter syndrome. Nat Genet. 2012;44:440–4.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24.
Srour M, Chitayat D, Caron V, Chassaing N, Bitoun P, Patry L, et al. Recessive and dominant mutations in retinoic acid receptor beta in cases with microphthalmia and diaphragmatic hernia. Am J Hum Genet. 2013;93:765–72.
Rainger J, Pehlivan D, Johansson S, Bengani H, Sanchez-Pulido L, Williamson KA, et al. Monoallelic and biallelic mutations in MAB21L2 cause a spectrum of major eye malformations. Am J Hum Genet. 2014;94:915–23.
Kalaskar VK, Alur RP, Li LK, Thomas JW, Sergeev YV, Blain D, et al. High-throughput custom capture sequencing identifies novel mutations in coloboma-associated genes: mutation in DNA-binding domain of retinoic acid receptor beta affects nuclear localization causing ocular coloboma. Hum Mutat. 2019;41:678–95.
Patel N, Khan AO, Alsahli S, Abdel-Salam G, Nowilaty SR, Mansour AM, et al. Genetic investigation of 93 families with microphthalmia or posterior microphthalmos. Clin Genet. 2018;93:1210–22.
Robitaille JM, Zheng B, Wallace K, Beis MJ, Tatlidil C, Yang J, et al. The role of Frizzled-4 mutations in familial exudative vitreoretinopathy and Coats disease. Brit J Ophthalmol. 2011;95:574–9.
Bang I, Kim HR, Beaven AH, Kim J, Ko SB, Lee GR, et al. Biophysical and functional characterization of Norrin signaling through Frizzled4. Proc Natl Acad Sci USA. 2018;115:8787–92.
Xu Q, Wang Y, Dabdoub A, Smallwood PM, Williams J, Woods C, et al. Vascular development in the retina and inner ear: control by Norrin and Frizzled-4, a high-affinity ligand-receptor pair. Cell. 2004;116:883–95.
Acknowledgements
We acknowledge generous support from the patients and their families as well as Claire Jeanton-Scaramouche for her precious help.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Aubert-Mucca, M., Pernin-Grandjean, J., Marchasson, S. et al. Confirmation of FZD5 implication in a cohort of 50 patients with ocular coloboma. Eur J Hum Genet 29, 131–140 (2021). https://doi.org/10.1038/s41431-020-0695-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-020-0695-8
