
Abstract
Although over 50 genes are known to cause renal malformation if mutated, the underlying genetic basis, most easily identified in syndromic cases, remains unsolved in most patients. In search of novel causative genes, whole-exome sequencing in a patient with renal, i.e., crossed fused renal ectopia, and extrarenal, i.e., skeletal, eye, and ear, malformations yielded a rare heterozygous variant in the GDF6 gene encoding growth differentiation factor 6, a member of the BMP family of ligands. Previously, GDF6 variants were reported to cause pleiotropic defects including skeletal, e.g., vertebral, carpal, tarsal fusions, and ocular, e.g., microphthalmia and coloboma, phenotypes. To assess the role of GDF6 in the pathogenesis of renal malformation, we performed targeted sequencing in 193 further patients identifying rare GDF6 variants in two cases with kidney hypodysplasia and extrarenal manifestations. During development, gdf6 was expressed in the pronephric tubule of Xenopus laevis, and Gdf6 expression was observed in the ureteric tree of the murine kidney by RNA in situ hybridization. CRISPR/Cas9-derived knockout of Gdf6 attenuated migration of murine IMCD3 cells, an effect rescued by expression of wild-type but not mutant GDF6, indicating affected variant function regarding a fundamental developmental process. Knockdown of gdf6 in Xenopus laevis resulted in impaired pronephros development. Altogether, we identified rare heterozygous GDF6 variants in 1.6% of all renal anomaly patients and 5.4% of renal anomaly patients additionally manifesting skeletal, ocular, or auricular abnormalities, adding renal hypodysplasia and fusion to the phenotype spectrum of GDF6 variant carriers and suggesting an involvement of GDF6 in nephrogenesis.
Similar content being viewed by others


Introduction
Structural defects of the kidney range from renal agenesis, hypoplasia, and dysplasia to duplication and fusion phenotypes, such as horseshoe kidneys and crossed fused renal ectopia. The latter is a rare form of renal anomaly where two fused kidneys come to lie on the same side of the spine, each with their own ureter, one of which crossing the midline to enter the bladder on the contralateral side. As other renal anomalies are also frequently associated with malformations of the urinary tract, such as ureteropelvic or ureterovesical junction obstruction with hydroureter, or vesicoureteral reflux (VUR), the term congenital anomalies of the kidney and urinary tract (CAKUT) has been coined to subsume these abnormalities. Taken together, CAKUT phenotypes account for 15–30% of all prenatally detected congenital malformations [1], and cause around 40% of cases with end-stage kidney disease in children and adolescents [2], thus representing a significant health burden. In around 85% of patients, CAKUT occur sporadically, while in the remaining 15% of cases familial occurrence is observed. CAKUT may occur in isolation or be part of a mild or complex syndromal disease. Since over 500 syndromes have been associated with CAKUT [3], it is not surprising that one-third of patients are additionally affected by extrarenal manifestations [4], and that around 20% of patients may have a genetic disorder that is not detected based on standard clinical evaluation [5]. Although over 50 genes are known to cause CAKUT in humans if mutated [6, 7], <20% of CAKUT manifestations can be explained by aberrations in these genes [8, 9], indicating a high genetic heterogeneity underlying these defects and making clear the need to identify new genes associated with renal development and malformation. However, the identification of causative genetic variants in cohorts of sporadic CAKUT patients and CAKUT families is hampered by variable expressivity, meaning that individuals harboring the same variant may have very different phenotypes within the broad CAKUT spectrum, and by incomplete penetrance implying that variant carriers may exist that are not affected by a CAKUT phenotype at all.
In the last few years, whole-exome sequencing (WES) using next generation sequencing (NGS) techniques was successfully applied to the study of germline variation underlying human CAKUT [10]. Thereby, novel CAKUT-associated genes were identified e.g., by using a linkage-based strategy in large CAKUT families [11, 12], a double hit-based strategy in smaller CAKUT families [13], an overlapping strategy in a cohort of patients with similar phenotypes [14], and a trio-based de novo strategy in patients with sporadic CAKUT [15, 16]. In addition, WES has improved the diagnostic yield of genetic CAKUT causes, particularly in syndromic cases [9].
Here, in an effort to identify new genes associated with renal malformation in humans, we used a WES approach to determine the genetic variation underlying renal anomalies in an index patient with syndromic CAKUT. By a targeted mutational screen in a cohort of 193 further patients with renal anomalies, expression analyses, and functional studies in a cellular system modified by CRISPR/Cas9 genome engineering and an animal model, we suggest that the candidate gene identified in the index patient, GDF6, plays a role in kidney development and malformation.
Subjects and methods
Patients
This study was conducted in accordance with the Declaration of Helsinki and approved by the Ethics Boards of Hannover Medical School, Hannover, Germany, Oslo University Hospital, Oslo, Norway, and University Children’s Hospital, Skopje, North Macedonia. Each family provided informed consent for participation in the study. A total of 194 patients with renal anomalies comprising 122 males and 72 females with a mean age of 10 years (range 1–35 years) were analyzed. Renal phenotypes of all 194 patients are listed in Supplementary Table 1. Rare heterozygous GDF6 variants predicted to be disease causing were detected in families F006, H435, and N038.
Family F006
The index patient, F006.II.1, born as the second daughter of non-consanguineous German parents is now 3 years old. After birth, renal ultrasound was notable for left-sided crossed fused renal ectopia (Fig. 1a). Voiding cystourethrography revealed two megaureters, both with orthotopic ostia in the bladder, one connected to the superior pelvis, and the other to the inferior pelvis of the left-sided fused kidneys (Fig. 1b), and grade-IV VUR in both ureters. Recurrent urinary tract infections were diagnosed. The patient also showed a left-convex torsion scoliosis, malformations of multiple vertebral bodies of the cervical and thoracic spine including butterfly and fused vertebrae, and a missing fifth sacral vertebral body and coccyx (Fig. 1c, d). A tethered cord was diagnosed because of a low standing conus medullaris, and detethering surgery was performed. The patient also presented with anal atresia and a rectovestibular fistula, which was surgically corrected. By echocardiography, two small muscular ventricular septal defects and a patent foramen ovale were diagnosed. Ophthalmologic examination revealed anisometropia with hyperopia, astigmatism, amblyopia, suspected microphthalmia, corneal opacities, and a best-corrected visual acuity of 0.16 in the left eye and of 1.0 in the right eye. In addition, the patient presented with a left-sided auricle dysplasia and aplasia of the external auditory canal, while the cochleae and the semicircular canals were unremarkable on both sides according to cranial MRI. Neurological examination was normal and no developmental or intellectual deficits were observed. Her 6-year-old sister and 36-year-old mother presented with right-sided preauricular pits, while renal ultrasound examinations revealed no abnormalities. Her father was clinically unremarkable and renal sonography was normal.

a Renal ultrasound of index patient F006.II.1 was notable for left-sided crossed fused kidney (K) ectopia with megaureters (U). b Voiding cystourethrography of patient F006.II.1 revealed grade-IV vesicoureteral reflux into two megaureters (U) with orthotopic ostia in the bladder (B) and connected to the superior or inferior dilated kidney (K) pelvis of the fused kidneys. c, d MRI of the spine of patient F006.II.1 showing malformations of cervical and thoracic vertebral bodies, e.g., fused and butterfly vertebrae (arrows) (c) and missing fifth sacral vertebral body and coccyx (arrow) (d). e, f Renal ultrasound of patient H435.II.1 at the age of 11 years was unremarkable on the right side (e), while the left kidney was hypodysplastic (f) and nonfunctional on DMSA scan. g Renal ultrasound of patient N038.II.1 at age 6 months showing renal hypodysplasia, as indicated by reduced size, hyperechogenicity, and reduced corticomedullary differentiation of the right kidney depicted. The left kidney was also hypodysplastic (not shown), and the patient required kidney transplantation at 5 years of age. h–j Pedigrees of families F006 (h), H435 (i), and N038 (j) with colored shading indicating phenotypical overlap with respect to renal, skeletal, auricular, and ocular anomalies in individuals with rare GDF6 variants. The GDF6 mutational status (V1: c.746C>A p.(Ala249Glu), V2: c.112G>C p.(Gly38Arg), WT: wild-type) is indicated, and corresponding electropherograms are shown for all analyzed family members (no DNA sample was available from individual N038.I.2). Clinical and radiological information was not available from individuals H435.II.3 (1 year of age) or N038.I.2. L, liver; the kidneys are marked by dotted lines (e–g).
Family H435
Twelve-year-old patient H435.II.1 was born as the second son of non-consanguineous Macedonian parents. After birth, the boy presented with a right-sided normal kidney and a left-sided hypodysplastic kidney, a diagnosis confirmed by ultrasound at age 11 years (Fig. 1e, f). The left kidney could not be visualized by a radionuclide scan using DMSA indicating that it is not functional. In addition, the patient showed a mild torsion scoliosis. The philtrum appeared shorter than usual, a high-arched palate and malocclusion due to prognathism were noted. His ears were of normal size but had a slight lop deformity. Except for the presence of a short frenulum, external genitalia were unremarkable. No developmental or intellectual deficits were noted. His 15-year-old brother also had high-arched palate and lop ears, whereby particularly the right ear was smaller and showed a poorly developed antihelix. A triangular-shaped chin was observed. Neurological examination was normal and no developmental or intellectual deficits were diagnosed. His 1-year-old sister was clinically unremarkable, but renal ultrasound or other examinations were not performed. His father presented with short stature. Renal sonography of both parents was normal.
Family N038
Fourteen-year-old patient N038.II.1 is the only daughter of an African mother and a Caucasian father. She was prenatally diagnosed with oligohydramnios and small kidneys. Renal ultrasound at age 6 months showed bilateral renal hypodysplasia as indicated by reduced size, hyperechogenicity, and reduced corticomedullary differentiation of the kidneys (right kidney shown in Fig. 1g). After birth, she additionally presented with macrocephaly, high-arched palate, and short narrow palpebral fissures. She required kidney transplantation at the age of 5 years and was re-transplanted at 9 years of age due to chronic rejection and noncompliance with drug treatment. At 14 years, she has poor kidney function and several comorbidities such as obesity and hypertension.
Animals
Husbandry and treatment of Xenopus laevis were approved by the Regierungspräsidium Freiburg, Germany. Mice were kept in accordance with the National Institutes of Health guidelines for the care and use of laboratory animals. All experiments on mice were approved by the Ethics Board of the Lower Saxony State Office for Consumer Protection and Food Safety. Murine embryos for gene expression analysis were derived from matings of wild-type mice with NMRI background. For timed pregnancies, vaginal plugs were checked in the morning after mating, and noon was defined as embryonic day (E) 0.5. Embryos were dissected in phosphate-buffered saline (PBS; Merck, Darmstadt, Germany) and fixed in 4% paraformaldehyde in PBS followed by dehydration using increasing methanol concentrations, i.e., incubation in 25%, 50%, and 75% methanol for 1 h each. Fixed embryos were stored in 100% methanol at −20 °C prior to in situ hybridization analyses.
WES and targeted GDF6 sequencing
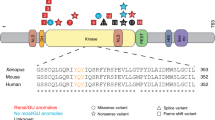
WES was performed on whole-blood DNA of one patient–sibling–parents index family, 30 additional patients with renal malformations, and 74 control individuals using the SureSelectXT Human All Exon V4 target enrichment kit (Agilent, Santa Clara, CA, USA) on a HiSeq 2000 (Illumina, San Diego, CA, USA) sequencer or the SureSelectXT Human All Exon V5+UTRs target enrichment kit (Agilent) on a HiSeq 2500 (Illumina) sequencer. All samples were sequenced to a mean target coverage of >50×. Sequencing data were aligned to the human reference genome (hg19) using the Biomedical Genomics Workbench (Qiagen, Hilden, Germany). WES data of the index family were annotated and prioritized using Ingenuity Variant Analysis (Qiagen) and our in-house NGS data analysis workflow as described in “Results” and summarized in Supplementary Tables 2 and 3. Using conventional chain termination protocols and a 3130XL Genetic Analyzer (Thermo Fisher Scientific, Waltham, MA, USA), all coding exons and adjacent intronic regions of GDF6 were analyzed for sequence variants in 163 further patients with kidney anomalies, selected GDF6 variants identified by WES were verified, and familial segregation analysis was done (oligonucleotide sequences are given in Supplementary Table 4). Nucleotide numbering of the identified variants reflects the nucleotide position in the coding sequence of human GDF6 mRNA (https://www.ncbi.nlm.nih.gov/nuccore/NM_001001557.4) (Supplementary Fig. 1).
Immunohistochemistry, RNA in situ hybridization, CRISPR/Cas9 genome engineering and cellular assays, knockdown and rescue experiments in Xenopus laevis
Procedures are described in “Supplementary materials” (including Supplementary Tables 5–8).
Statistical analysis
Statistical analysis was done using MATLAB and Statistics Toolbox Release 2018b (The MathWorks, Inc., Natick, MA, USA). Student’s t-test or Fisher’s exact test were used, as applicable, and p values are indicated (*p < 0.05, **p < 0.01, and ***p < 0.001).
Results
Using WES, a rare heterozygous GDF6 missense variant, c.746C>A p.(Ala249Glu), was detected in the index patient
Under the assumption that NGS techniques are particularly successful in identifying the genetic cause in patients with syndromic CAKUT, we applied WES to whole-blood DNA of female patient F006.II.1 with a renal malformation (i.e., crossed fused renal ectopia) as well as skeletal (e.g., scoliosis, fused and butterfly vertebrae) (Fig. 1a–d), auricular (i.e., auricle dysplasia and aplasia of the external auditory canal), ocular (e.g., anisometropia), and other extrarenal anomalies, and of her mother, father, and sister who were not affected by renal anomalies. As no rare de novo, homozygous, or compound-heterozygous variants predicted to be disease causing could be detected in the high-quality exome data of patient F006.II.1, variants were prioritized using the strategy summarized in Supplementary Table 2. Prioritization of high-quality variants of patient F006.II.1 by seriousness, rareness, exclusiveness, and localization in genes (n = 207) reported to be mutated in at least one patient with syndromic CAKUT according to our in-house gene list yielded five rare (minor allele frequency (MAF) ≤ 1%) non-silent variants not present in controls and predicted to be disease causing by at least one prediction tool (MutationTaster, SIFT, or PolyPhen-2) (Supplementary Tables 2 and 3). One of these five variants, the GDF6 variant c.746C>A p.(Ala249Glu), was presumed causative in patient F006.II.1 because it is reported to be disease causing by the HGMD Professional (v2018.2; Qiagen) and ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) databases in patients with skeletal and ocular anomalies [17,18,19,20,21,22] matching the patient’s extrarenal phenotype (Supplementary Table 9). By direct sequencing, the GDF6 c.746C>A variant was confirmed to be heterozygous in the patient as well as in her sister and mother, both affected by the same mild auricular anomaly (Fig. 1h). The c.746C>A variant is located in the second exon of GDF6 and occurs with a MAF of 0.001948 in the global population cohort and a MAF of 0.003783 in the non-Finnish European cohort of the Genome Aggregation Database (gnomAD v2.1.1) (Table 1). The amino acid alanine at position 249 is evolutionary conserved and located in the propeptide/prodomain of GDF6 (Supplementary Fig. 1). Based on the ACMG/AMP 2015 guidelines [23], we classified the c.746C>A variant as “pathogenic” (Table 1).
Heterozygous GDF6 missense variants were detected in a total of three of 194 patients (1.6%) with renal malformations and three of 56 patients (5.4%) with renal plus skeletal, ocular or auricular anomalies
Having identified a GDF6 variant previously associated with skeletal and ocular anomalies in a patient presenting with these phenotypes and additionally with a renal malformation, we explored the frequency of rare GDF6 variants in a cohort of patients with renal anomalies. A total of 193 additional cases were either analyzed by WES (30 patients) or targeted sequencing of the GDF6 gene (163 patients). By targeted sequencing, we identified the GDF6 variant c.746C>A in an additional patient, H435.II.1 from North Macedonia, presenting with a hypodysplastic left kidney (Fig. 1f, i) that was nonfunctional on DMSA scan, mild skeletal, and auricular phenotypes, i.e., torsion scoliosis, high-arched palate, and lop ears (Table 1). The GDF6 c.746C>A variant was detected in a heterozygous state in the patient, his father who presented with short stature, his brother who had a high-arched palate and lop ears, and a 1-year-old sister who was not further examined (Fig. 1i). Another rare heterozygous GDF6 missense variant, c.112G>C p.(Gly38Arg), was detected by targeted sequencing in female patient N038.II.1 presenting with bilateral renal hypodysplasia (Fig. 1g) requiring kidney transplantation at age 5 years in addition to skeletal, i.e., macrocephaly and high-arched palate, and ocular, i.e., short narrow palpebral fissures, phenotypes (Fig. 1j and Table 1). As the c.112G>C variant was not inherited from her Caucasian father, it may be a de novo variant or have been inherited from her African mother (Fig. 1j) from whom no DNA sample was available for clarification. Located in the first exon of GDF6 (Supplementary Fig. 1), the c.112G>C variant occurs with a MAF of 0.000656 in the global population cohort, a MAF of 0.007016 in the African population cohort, and is not present in the non-Finnish European cohort of the gnomAD database v2.1.1 (Table 1). While the c.112G>C variant was of “uncertain significance” based on the ACMG/AMP 2015 guidelines [23], the prediction tools SIFT and PolyPhen-2 rated it as “damaging” or “possibly damaging” (Table 1). The amino acid glycine at position 38 is evolutionary highly conserved. In total, we detected rare heterozygous GDF6 missense variants predicted to be disease causing in 3 of 194 families with renal anomalies (1.6%). Clinical or radiological reevaluation of the skeleton, eye or ear revealed skeletal, ocular, or auricular abnormalities in 56 of the 194 patients with renal anomalies analyzed. As the three GDF6 variant carriers were among these patients, 3 of 56 patients (5.4%) with renal plus skeletal, ocular, or auricular features carried GDF6 variants with a disease-causing prediction, a significant frequency increase compared with that in renal anomaly patients without abnormalities of the skeleton, eye, or ear (0/138, p = 0.0231, Fisher’s exact test).
GDF6 is expressed in the infant human kidney, and in the developing murine urogenital system and pronephros of Xenopus laevis
Having detected rare GDF6 variants in patients with renal anomalies, we went on to explore a possible role for GDF6 in kidney development by determining the expression pattern of GDF6 in the infant human kidney as well as in Mus musculus and Xenopus laevis during development. GDF6 protein expression was detected in the human infant kidney, most prominently in proximal tubules, by immunohistochemistry (Fig. 2a). During lower vertebrate development, expression of gdf6 was observed in the area of the pronephros, the embryonic kidney of Xenopus laevis, with particular enhancement in the tissue just adjacent to the pronephric tubule at stage 38 by whole-mount RNA in situ hybridization. Expression at this stage was also detected around the eye vesicle, branchial arches, notochord, and neural tube of Xenopus laevis embryos (Fig. 2b). During murine urogenital system development, Gdf6 mRNA was present in the ureteric tips at E11.5 shortly after onset of metanephros development. At E13.5, expression of Gdf6 in the kidney reached its peak and was found in all compartments, i.e., the ureter and ureteric tips, of the developing ureteric tree. After onset of collecting duct differentiation at E14.5, Gdf6 transcript levels decreased in the kidney, and renal expression was barely detectable at E18.5. At E11.5, Gdf6 was also detectable in the mesothelial lining of the abdominal cavity and weakly in the epithelium of the urogenital sinus, which develops into the bladder. Expression in the bladder urothelium vanished after E14.5. Gdf6 mRNA was also found in the nephric duct or differentiated vas deferens of male embryos at all analyzed stages (Fig. 2c, data partly not shown).

a By immunohistochemical detection of GDF6 and the marker proteins aquaporin-1 (AQP1, straight and convoluted proximal tubule and thin descending limb of loop of Henle), aquaporin-2 (AQP2, collecting duct), and Tamm-Horsfall protein (THP, thick ascending limb of loop of Henle and distal tubule) in a normal human infant kidney section, GDF6 localized most prominently to proximal tubules. b By whole-mount RNA in situ hybridization in Xenopus laevis at stage 38, gdf6 was expressed in the tissue surrounding the pronephric tubule (arrow, enlarged image) as well as in the developing eye, branchial arches, notochord, and neural tube. c RNA in situ hybridization analysis on sagittal sections of the murine kidney and bladder from E11.5 to E18.5. Gdf6 expression was present in all compartments of the developing ureteric tree, including the ureteric tips (ut) and the ureter (u), at E11.5 to E14.5, dropped after onset of collecting duct differentiation, and was barely detectable at E18.5 (p pelvis, c cortex). In the lower urogenital tract, Gdf6 expression was found in the mesothelial lining (m) of the abdominal cavity at E11.5, in the undifferentiated urothelium of the urogenital sinus (us) and the bladder (bl) until E14.5, and in the nephric duct (nd) of male embryos at all analyzed stages (data partly not shown). Sections from three independent murine specimens were analyzed.
Knockout of Gdf6 in murine inner medullary collecting duct (mIMCD3) cells impairs cell migration, an effect rescued by expression of wild-type not mutant GDF6 in Gdf6−/− mIMCD3 cells
Next, we generated an in vitro test system to determine whether GDF6 impacts cell migration, a central process in development, and to assess whether the identified GDF6 variants affect this function. Using CRISPR/Cas9 technology, a single guide RNA targeting the first coding exon of Gdf6 was applied to knockout Gdf6 in mIMCD3 cells. Two Gdf6−/− mIMCD3 cell clones with frameshift variants predicted to result in premature stop codons and nonfunctional proteins were identified, i.e., clone 32 harboring the homozygous Gdf6 variant c.377_378delCA p.(Ser126Cysfs*2), and clone 34 containing the biallelic Gdf6 variants c.373_376delAAGT p.(Lys125Glnfs*9) and c.377_378delCA p.(Ser126Cysfs*2) (Supplementary Fig. 2). A mIMCD3 cell clone with no mutational event in Gdf6 (clone 2, Gdf6+/+) was also identified and used as a control (Supplementary Fig. 2). No differences in cell viability were observed when comparing mIMCD3 cells and Gdf6+/+ mIMCD3 cell clone 2 with the Gdf6−/− mIMCD3 cell clones 32 or 34 using a cell viability assay (Supplementary Fig. 3).
A time series analyzing mIMCD3 cell migration in a wound healing assay resulted in a reduction of the cell-free area by 50% after 8 h (Supplementary Fig. 4). Therefore, in subsequent analyses the cell-free areas were determined and compared at 0 h and 8 h. While migration of mIMCD3 cells and Gdf6+/+ mIMCD3 cell clone 2 did not differ significantly, migration of Gdf6−/− mIMCD3 cell clones 32 and 34 was significantly decreased compared with Gdf6+/+ mIMCD3 cell clone 2 (p = 0.002 and p = 0.005, respectively; Fig. 3a), thereby providing evidence that Gdf6 knockout impacts cell migration. Migration was also significantly impaired in Gdf6+/− mIMCD3 cell clone 30 versus Gdf6+/+ mIMCD3 cell clone 2 (p = 0.041; Supplementary Fig. 5), indicating an effect on migration of heterozygously mutated cells, a cellular model for patients with heterozygous GDF6 variants. A significant increase in migration of Gdf6−/− mIMCD3 cell clone 32 stably transfected with a wild-type GDF6 expression construct was detected compared with Gdf6−/− mIMCD3 cell clone 32 transfected with empty vector (p = 0.007; Fig. 3b and Supplementary Fig. 6), showing that re-expression of human wild-type GDF6 can rescue the reduced migration. In contrast, migration of Gdf6−/− mIMCD3 cell clone 32 transfected with constructs expressing GDF6 variants c.112G>C or c.746C>A versus GDF6 wild-type was significantly reduced (p = 0.003 and p = 0.008, respectively; Fig. 3b and Supplementary Fig. 6), demonstrating that the identified GDF6 variants are not functional in this assay.

a Relative to mIMCD3 cells, migration of Gdf6−/− mIMCD3 cells (clones 32 and 34) was significantly decreased compared with Gdf6+/+ mIMCD3 cell clone 2. b The effect of Gdf6 knockout in mIMCD3 cell clone 32 expressing empty vector (vector control) relative to mIMCD3 cells was partially rescued by stable expression of human wild-type (WT) GDF6 in mIMCD3 cell clone 32. Conversely, relative cell migration of Gdf6−/− mIMCD3 cell clone 32 stably expressing GDF6 c.112G>C or c.746C>A variants was significantly reduced compared with Gdf6−/− mIMCD3 cell clone 32 stably expressing wild-type GDF6. All results are mean ± SD from three independent experiments. Scale bar: 150 µm. N.s. not significant; **p < 0.01; ***p < 0.001.
Morpholino (MO) knockdown of gdf6 in Xenopus laevis impairs pronephros development
To explore a possible role of gdf6 in renal development in vivo, we performed gdf6 knockdown unilaterally using a specific gdf6 antisense MO in Xenopus laevis tadpoles, and analyzed the developing pronephros, constituting the embryonic kidney in lower vertebrates, at stage 39 (Fig. 4). The pronephric area, calculated as log2 ratio of the injected and uninjected side of the tadpole, was significantly reduced after gdf6 MO injection compared with control MO injection (p = 7.6 × 10−13; Fig. 4). This effect was partially rescued by co-injection of GDF6 mRNA with gdf6 MO that increased the pronephros area significantly compared with injecting gdf6 MO alone (p = 0.035; Fig. 4). Thereby, we demonstrate a role for gdf6 in pronephric tubule development in Xenopus laevis.

Xenopus laevis stage (st.) 39 tadpoles were unilaterally injected with a control MO or a translation-blocking gdf6 MO with or without co-injection of GDF6 mRNA, and stained with fluorescein-labeled lectin to visualize the pronephros. The ratios of the pronephros areas of the injected (inj.) and the uninjected (uninj.) side of the embryo were calculated and log2 transformed. Knockdown with the gdf6 MO led to a significantly reduced pronephros area. This effect was significantly rescued by the co-injection of GDF6 mRNA. Results are mean ± SEM from n embryos analyzed in four independent experiments. N.s. not significant; *p < 0.05; ***p < 0.001.
Discussion
In the present study systematically investigating a role of GDF6 in renal anomalies, we identified rare heterozygous GDF6 variants in 1.6% of patients with kidney malformations. Initially, we found GDF6 to be associated with renal malformation by an unbiased screen for germline variation in a patient with renal as well as skeletal, auricular, ocular, and other anomalies. This patient had been chosen for WES analysis because NGS technologies have been particularly successful in identifying causative genes in syndromic CAKUT [9]. Subsequently, we detected two further renal anomaly patients with rare heterozygous GDF6 variants among the 193 patients additionally analyzed. In line with the extrarenal manifestations of our three renal anomaly patients carrying GDF6 variants, variants in GDF6 have previously been reported in patients with skeletal phenotypes, i.e., (i) Klippel–Feil syndrome (KFS) with vertebral segmentation defects frequently associated with scoliosis, rib abnormalities, and Sprengel’s deformity now known as KFS1 [17, 18], (ii) Chiari malformations [17, 24], and (iii) multiple synostoses syndrome including carpal and tarsal fusions [25,26,27], as well as ocular phenotypes, i.e., (i) microphthalmia [20,21,22], (ii) coloboma [18, 28], (iii) Leber congenital amaurosis or juvenile retinitis pigmentosa [19], and (iv) glaucoma [29]. By adding three patients with renal anomalies to the two patients previously reported [17, 18], a total of 5 of 86 (5.8%) individuals carrying rare GDF6 variants described here and in the literature (Supplementary Table 9) are known to be affected by congenital kidney malformations. This finding suggests that renal sonography is warranted in patients carrying rare GDF6 variants irrespective of the other abnormalities or disorders they may present with. Vice versa, GDF6 mutational analysis may be advisable in patients with renal anomalies, in particular in those cases additionally affected by the skeletal and ocular phenotypes previously associated with rare GDF6 variants and also present in the renal anomaly patients with GDF6 variants of this study. Accordingly, the percentage of rare GDF6 variants in patients with renal plus bone, eye, or ear abnormalities was 5.4%, significantly higher than in renal anomaly patients without these extrarenal manifestations. In this context, it is notable that we detected one GDF6 variant, the missense variant c.746C>A, recurrently in renal anomaly patients, resulting in a significant frequency increase compared with the cohort of the 1000 Genomes Project. Previously, the GDF6 c.746C>A variant was reported in patients with KFS-like skeletal anomalies [17, 18, 20], Chiari malformation [24], micro- or anophthalmia [20,21,22], and Leber congenital amaurosis or juvenile retinitis pigmentosa [19] (Supplementary Table 9), demonstrating that it can be associated with a spectrum of different phenotypes. In line with these findings, we detected skeletal anomalies in four of seven GDF6 c.746C>A variant carriers from two families, and ocular anomalies in one carrier. Similar to kidney malformations that were present in two of the seven GDF6 c.746C>A variant carriers here, penetrance is reduced for these abnormalities. Reduced penetrance is a known feature associated with renal anomalies. This is exemplified by a family with Stickler syndrome and a heterozygous nonsense variant in the BMP4 gene encoding bone morphogenetic protein 4, a ligand related to GDF6, with renal dysplasia in only one of five family members carrying the BMP4 variant [30].
GDF6 encodes growth differentiation factor 6, a member of the BMP family within the transforming growth factor beta (TGF-β) superfamily of ligands that utilize type I and type II transmembrane serine–threonine kinase receptors [31]. Therefore, in GDF6 variant carriers reduced penetrance for renal malformations may be explained by the complexity of TGF-β signaling comprising numerous ligands (around 30), receptors, and downstream interacting proteins [31], suggesting some redundancy. The GDF6 amino acid sequence consists of three domains. These are a signal peptide, a propeptide/prodomain, and a mature receptor-binding carboxy-terminal TGF-domain that is highly conserved between species, i.e., it is more than 90% identical in Xenopus laevis and mouse [32]. Interestingly, 74% of the GDF6 variants detected in patients with different phenotypes (Supplementary Table 9), including the recurrent c.746C>A variant and the c.112G>C variant identified in renal anomaly patients here, affect amino acids located in the prodomain of the GDF6 sequence. Although it is less conserved, the prodomain of TGF-β superfamily ligands is known to regulate the synthesis, extracellular localization, and activity of these proteins [33]. The prodomain of BMP4, another member of the BMP family of ligands, for instance, is necessary to generate stable BMP4/7 heterodimers with enhanced bioactivity in vivo [34]. Therefore, it is not unexpected that although the c.746C>A variant affects an amino acid located in the prodomain of GDF6, the activity of variant GDF6 was significantly decreased in a SOX9-reporter luciferase assay [18], and amounts of variant pre-pro-protein and mature ligand were reduced in the media of transfected cells [19] compared with wild-type. Furthermore, we show here that impaired cell migration of murine Gdf6 knockout cells from the renal inner medullary collecting duct is rescued by expression of wild-type GDF6 but not of c.746C>A and c.112G>C variants, indicating that both are hypomorphic variants with respect to cell movements, a fundamental process in development.
BMP signaling is highly implicated in embryogenesis including nephrogenesis and development [35,36,37,38]. CAKUT phenotypes were observed with high penetrance in mice carrying a heterozygous or homozygous knockout for genes encoding BMP ligands such as BMP4 [39], BMP7 [40], and GDF11 [41]. Pleiotropic defects have been described as a result of Gdf6 knockout, knockdown, or variation in mice, zebrafish, or Xenopus laevis including defects in joint, ligament, and cartilage formation causing carpal and tarsal fusions and coronal craniosynostosis [42], altered tail tendon fascicles [43], shorter lengths of digits and dermal flat bones in the skull [44], microphthalmia, anophthalmia, and coloboma [20, 45]. Whether urogenital tract anomalies exist has not previously been examined in these animal models. Based on the results from our expression and functional studies and the finding that other BMP ligands, such as BMP4, regulate the budding site and elongation of the developing mouse ureter [39], we propose that the rare GDF6 variants detected in five renal anomaly patients here and previously [17, 18] may be causally related to their kidney malformation. This proposal is in line with the spectrum of renal abnormalities seen in GDF6 variant carriers that includes renal agenesis [17] and renal hypodysplasia (two cases here), two anomalies also found in patients carrying BMP4 variants [46]. Kidney agenesis or hypoplasia are potentially caused by defects in ureteric budding and branching of the ureteric tree induced by abberant BMP signaling as detected in Bmp4 heterozygous knockout mice [39]. It is quite conceivable that such budding and branching defects giving rise to missing or small dysplastic kidneys also occur in human carriers of rare GDF6 variants because the developing ureteric tree expresses Gdf6, and migration of collecting duct cells that originate from the ureteric tree is impaired by Gdf6 knockout, as shown here. Similarly, pronephros size was reduced by gdf6 knockdown in Xenopus laevis. We and others additionally observed kidney abnormalities involving renal fusions, i.e., a horseshoe kidney [18] and a crossed fused renal ectopia (one case here), in patients with rare GDF6 variants, similar to the skeletal fusions seen (Supplementary Table 9). These data imply that BMP signaling is also linked to skeletal and renal fusions, similar to observations in a conditional Bmp4 knockout mouse in which reduced BMP signaling resulted in hindlimb fusion [47]. According to an established view, renal fusions result as a consequence of abnormal renal ascent during embryogenesis [48], possibly connecting GDF6 to the migration of the kidneys as also suggested by our cell migration studies.
In summary, we identified rare heterozygous GDF6 variants in 1.6% of all patients of our renal anomaly cohort, and in 5.4% of those patients additionally manifesting skeletal, ocular, or auricular abnormalities suggesting that GDF6 is associated with human kidney malformations. The phenotype spectrum identified in renal anomaly patients with GDF6 variants ranged from hypodysplasia to fusion. Furthermore, Gdf6 expression in the murine developing ureteric tree, diminished migration of murine Gdf6−/− collecting duct cells and impaired pronephros development after gdf6 knockdown in Xenopus laevis may support a role of GDF6 in kidney development.
References
Queisser-Luft A, Stolz G, Wiesel A, Schlaefer K, Spranger J. Malformations in newborn: results based on 30,940 infants and fetuses from the Mainz congenital birth defect monitoring system (1990–1998). Arch Gynecol Obstet. 2002;266:163–7.
Harambat J, van Stralen KJ, Kim JJ, Tizard EJ. Epidemiology of chronic kidney disease in children. Pediatr Nephrol. 2012;27:363–73.
Limwongse C. Syndromes and malformations of the urinary tract. In: Avner E, Harmon W, Niaudet P, Yoshikawa N, editors. Pediatric nephrology. Berlin, Heidelberg, Germany: Springer; 2009. p. 121–56.
Stoll C, Dott B, Alembik Y, Roth MP. Associated nonurinary congenital anomalies among infants with congenital anomalies of kidney and urinary tract (CAKUT). Eur J Med Genet. 2014;57:322–8.
Sanna-Cherchi S, Westland R, Ghiggeri GM, Gharavi AG. Genetic basis of human congenital anomalies of the kidney and urinary tract. J Clin Invest. 2018;128:4–15.
Kosfeld A, Martens H, Hennies I, Haffner D, Weber RG. Kongenitale Anomalien der Nieren und ableitenden Harnwege (CAKUT). Med Genet. 2018;30:448–60.
van der Ven AT, Vivante A, Hildebrandt F. Novel insights into the pathogenesis of monogenic congenital anomalies of the kidney and urinary tract. J Am Soc Nephrol. 2018;29:36–50.
Heidet L, Moriniere V, Henry C, De Tomasi L, Reilly ML, Humbert C, et al. Targeted exome sequencing identifies PBX1 as involved in monogenic congenital anomalies of the kidney and urinary tract. J Am Soc Nephrol. 2017;28:2901–14.
van der Ven AT, Connaughton DM, Ityel H, Mann N, Nakayama M, Chen J, et al. Whole-exome sequencing identifies causative mutations in families with congenital anomalies of the kidney and urinary tract. J Am Soc Nephrol. 2018;29:2348–61.
Nigam A, Knoers N, Renkema KY. Impact of next generation sequencing on our understanding of CAKUT. Semin Cell Dev Biol. 2019;91:104–10.
Vivante A, Kleppa MJ, Schulz J, Kohl S, Sharma A, Chen J, et al. Mutations in TBX18 cause dominant urinary tract malformations via transcriptional dysregulation of ureter development. Am J Hum Genet. 2015;97:291–301.
Brophy PD, Rasmussen M, Parida M, Bonde G, Darbro BW, Hong X, et al. A gene implicated in activation of retinoic acid receptor targets is a novel renal agenesis gene in humans. Genetics. 2017;207:215–28.
Humbert C, Silbermann F, Morar B, Parisot M, Zarhrate M, Masson C, et al. Integrin alpha 8 recessive mutations are responsible for bilateral renal agenesis in humans. Am J Hum Genet. 2014;94:288–94.
De Tomasi L, David P, Humbert C, Silbermann F, Arrondel C, Tores F, et al. Mutations in GREB1L cause bilateral kidney agenesis in humans and mice. Am J Hum Genet. 2017;101:803–14.
Kosfeld A, Kreuzer M, Daniel C, Brand F, Schafer AK, Chadt A, et al. Whole-exome sequencing identifies mutations of TBC1D1 encoding a Rab-GTPase-activating protein in patients with congenital anomalies of the kidneys and urinary tract (CAKUT). Hum Genet. 2016;135:69–87.
Kosfeld A, Brand F, Weiss AC, Kreuzer M, Goerk M, Martens H, et al. Mutations in the leukemia inhibitory factor receptor (LIFR) gene and Lifr deficiency cause urinary tract malformations. Hum Mol Genet. 2017;26:1716–31.
Tassabehji M, Fang ZM, Hilton EN, McGaughran J, Zhao Z, Bock CEd, et al. Mutations in GDF6 are associated with vertebral segmentation defects in Klippel–Feil syndrome. Hum Mutat. 2008;29:1017–27.
Asai-Coakwell M, French CR, Ye M, Garcha K, Bigot K, Perera AG, et al. Incomplete penetrance and phenotypic variability characterize Gdf6-attributable oculo-skeletal phenotypes. Hum Mol Genet. 2009;18:1110–21.
Asai-Coakwell M, March L, Dai XH, Duval M, Lopez I, French CR, et al. Contribution of growth differentiation factor 6-dependent cell survival to early-onset retinal dystrophies. Hum Mol Genet. 2013;22:1432–42.
den Hollander AI, Biyanwila J, Kovach P, Bardakjian T, Traboulsi EI, Ragge NK, et al. Genetic defects of GDF6 in the zebrafish out of sight mutant and in human eye developmental anomalies. BMC Genet. 2010;11:102.
Gonzalez-Rodriguez J, Pelcastre EL, Tovilla-Canales JL, Garcia-Ortiz JE, Amato-Almanza M, Villanueva-Mendoza C, et al. Mutational screening of CHX10, GDF6, OTX2, RAX and SOX2 genes in 50 unrelated microphthalmia-anophthalmia-coloboma (MAC) spectrum cases. Br J Ophthalmol. 2010;94:1100–4.
Slavotinek AM, Garcia ST, Chandratillake G, Bardakjian T, Ullah E, Wu D, et al. Exome sequencing in 32 patients with anophthalmia/microphthalmia and developmental eye defects. Clin Genet. 2015;88:468–73.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24.
Markunas CA, Soldano K, Dunlap K, Cope H, Asiimwe E, Stajich J, et al. Stratified whole genome linkage analysis of Chiari type I malformation implicates known Klippel-Feil syndrome genes as putative disease candidates. PLoS ONE. 2013;8:e61521.
Drage Berentsen R, Haukanes BI, Juliusson PB, Rosendahl K, Houge G. A novel GDF6 mutation in a family with multiple synostoses syndrome without hearing loss. Mol Syndromol. 2019;9:228–34.
Terhal PA, Verbeek NE, Knoers N, Nievelstein R, van den Ouweland A, Sakkers RJ, et al. Further delineation of the GDF6 related multiple synostoses syndrome. Am J Med Genet A. 2018;176:225–9.
Wang J, Yu T, Wang Z, Ohte S, Yao R-E, Zheng Z, et al. A new subtype of multiple synostoses syndrome is caused by a mutation in GDF6 that decreases its sensitivity to noggin and enhances its potency as a BMP signal. J Bone Min Res. 2016;31:882–9.
Chassaing N, Causse A, Vigouroux A, Delahaye A, Alessandri J-L, Boespflug-Tanguy O, et al. Molecular findings and clinical data in a cohort of 150 patients with anophthalmia/microphthalmia. Clin Genet. 2014;86:326–34.
Huang X, Xiao X, Jia X, Li S, Li M, Guo X, et al. Mutation analysis of the genes associated with anterior segment dysgenesis, microcornea and microphthalmia in 257 patients with glaucoma. Int J Mol Med. 2015;36:1111–7.
Nixon TRW, Richards A, Towns LK, Fuller G, Abbs S, Alexander P, et al. Bone morphogenetic protein 4 (BMP4) loss-of-function variant associated with autosomal dominant Stickler syndrome and renal dysplasia. Eur J Hum Genet. 2019;27:369–77.
De Caestecker M. The transforming growth factor-b: superfamily of receptors. Cytokine Growth Factor Rev. 2004;15:1–11.
Chang C, Hemmati-Brivanlou A. Xenopus GDF6, a new antagonist of noggin and a partner of BMPs. Development. 1999;126:3347–57.
Harrison CA, Al-Musawi SL, Walton KL. Prodomains regulate the synthesis, extracellular localisation and activity of TGF-β superfamily ligands. Growth Factors. 2011;29:174–86.
Neugebauer JM, Kwon S, Kim HS, Donley N, Tilak A, Sopory S, et al. The prodomain of BMP4 is necessary and sufficient to generate stable BMP4/7 heterodimers with enhanced bioactivity in vivo. Proc Natl Acad Sci USA. 2015;112:E2307–16.
Cain JE, Hartwig S, Bertram JF, Rosenblum ND. Bone morphogenetic protein signaling in the developing kidney: present and future. Differentiation. 2008;76:831–42.
Nishinakamura R, Sakaguchi M. BMP signaling and its modifiers in kidney development. Pediatr Nephrol. 2014;29:681–6.
Wang RN, Green J, Wang Z, Deng Y, Qiao M, Peabody M, et al. Bone morphogenetic protein (BMP) signaling in development and human diseases. Genes Dis. 2014;1:87–105.
Mamo TM, Wittern AB, Kleppa M-J, Bohnenpoll T, Weiss A-C, Kispert A. BMP4 uses several different effector pathways to regulate proliferation and differentiation in the epithelial and mesenchymal tissue compartments of the developing mouse ureter. Hum Mol Genet. 2017;26:3553–63.
Miyazaki Y, Oshima K, Fogo A, Hogan BL, Ichikawa I. Bone morphogenetic protein 4 regulates the budding site and elongation of the mouse ureter. J Clin Invest. 2000;105:863–73.
Dudley AT, Lyons KM, Robertson EJ. A requirement for bone morphogenetic protein-7 during development of the mammalian kidney and eye. Genes Dev. 1995;9:2795–807.
Esquela AF, Lee SJ. Regulation of metanephric kidney development by growth/differentiation factor 11. Dev Biol. 2003;257:356–70.
Settle SH, Rountree RB, Sinha A, Thacker A, Higgins K, Kingsley DM. Multiple joint and skeletal patterning defects caused by single and double mutations in the mouse Gdf6 and Gdf5 genes. Dev Biol. 2003;254:116–30.
Mikic B, Rossmeier K, Bierwert L. Identification of a tendon phenotype in GDF6 deficient mice. Anat Rec. 2009;292:396–400.
Indjeian VB, Kingman GA, Jones FC, Guenther CA, Grimwood J, Schmutz J, et al. Evolving new skeletal traits by cis-regulatory changes in bone morphogenetic proteins. Cell. 2016;164:45–56.
Asai-Coakwell M, French CR, Berry KM, Ye M, Koss R, Somerville M, et al. GDF6, a novel locus for a spectrum of ocular developmental anomalies. Am J Hum Genet. 2007;80:306–15.
Weber S, Taylor JC, Winyard P, Baker KF, Sullivan-Brown J, Schild R, et al. SIX2 and BMP4 mutations associate with anomalous kidney development. J Am Soc Nephrol. 2008;19:891–903.
Suzuki K, Adachi Y, Numata T, Nakada S, Yanagita M, Nakagata N, et al. Reduced BMP signaling results in hindlimb fusion with lethal pelvic/urogenital organ aplasia: a new mouse model of sirenomelia. PLoS ONE. 2012;7:e43453.
Taghavi K, Kirkpatrick J, Mirjalili SA. The horseshoe kidney: Surgical anatomy and embryology. J Pediatr Urol. 2016;12:275–80.
Acknowledgements
We deeply appreciate the participation of patients and families in this study. We wish to thank Achim Gossler and Michael Klintschar, Hannover Medical School, Hannover, Germany for providing mIMCD3 cells and human infant kidney sections, respectively. The Xenopus laevis gdf6 construct for whole-mount RNA in situ hybridization was kindly provided by Alexandra Schambony, Friedrich-Alexander University Erlangen-Nuremberg, Erlangen, Germany. We wish to acknowledge the assistance of the Cell Sorting Core Facility of Hannover Medical School. Open Access Funding provided by Projekt DEAL.
Funding
This research was supported by grants from the Else Kröner-Fresenius-Stiftung to RGW (2014_A234), the Deutsche Forschungsgemeinschaft to AnC (KO5614/2-1) and SSL (LI1817/2-1), the Swiss National Science Foundation (SNF) to SSL (NCCR Kidney.CH), and the Hochschulinterne Leistungsförderung (HiLF) program of Hannover Medical School to FB. The Cell Sorting Core Facility of Hannover Medical School is supported by the Braukmann-Wittenberg-Herz-Stiftung and the Deutsche Forschungsgemeinschaft.
Author information
Authors and Affiliations
Contributions
HM, AnC, FB, DH, and RGW designed research; HM analyzed WES data, performed and provided figures of all experiments except RNA in situ hybridization and Xenopus studies, compiled literature data; HM, AnC, FB, and RGW analyzed these data; MG and SSL performed, analyzed, and provided figures of Xenopus studies; ACW and AK performed, analyzed, and provided figures of RNA in situ hybridization on murine tissue; IH, ACG, ZG, TS, VT, AB, and DH performed clinical examinations, contributed patient materials, data, and clinical expertise; ArC provided technical expertise; RG generated WES data; HM, AnC, FB, DH, and RGW wrote the manuscript; all authors reviewed and revised the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Martens, H., Hennies, I., Getwan, M. et al. Rare heterozygous GDF6 variants in patients with renal anomalies. Eur J Hum Genet 28, 1681–1693 (2020). https://doi.org/10.1038/s41431-020-0678-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-020-0678-9
