
Abstract
Nonsyndromic hearing loss is genetically heterogeneous. Despite comprehensive genetic testing, many cases remain unsolved because the clinical significance of identified variants is uncertain or because biallelic pathogenic variants are not identified for presumed autosomal recessive cases. Common synonymous variants are often disregarded. Determining the pathogenicity of synonymous variants may improve genetic diagnosis. We report a synonymous variant c.9861 C > T/p.(Gly3287=) in MYO15A in homozygosity or compound heterozygosity with another pathogenic or likely pathogenic MYO15A variant in 10 unrelated families with nonsyndromic sensorineural hearing loss. Biallelic variants in MYO15A were identified in 21 affected and were absent in 22 unaffected siblings. A mini-gene assay confirms that the synonymous variant leads to abnormal splicing. The variant is enriched in the Ashkenazi Jewish population. Individuals carrying biallelic variants involving c.9861 C > T often exhibit progressive post-lingual hearing loss distinct from the congenital profound deafness typically associated with biallelic loss-of-function MYO15A variants. This study establishes the pathogenicity of the c.9861 C > T variant in MYO15A and expands the phenotypic spectrum of MYO15A-related hearing loss. Our work also highlights the importance of multicenter collaboration and data sharing to establish the pathogenicity of a relatively common synonymous variant for improved diagnosis and management of hearing loss.
Similar content being viewed by others


Introduction
Hearing loss is a genetically heterogenous condition. An autosomal recessive inheritance pattern is most common and accounts for 70% of genetic deafness [1]. To date, more than 100 genomic loci and at least 70 genes have been implicated in autosomal recessive nonsyndromic deafness (designated as DFNB) (Van Camp G, Smith RJH. Hereditary Hearing Loss Homepage; https://hereditaryhearingloss.org).
Comprehensive genetic testing using high-throughput DNA sequencing technology has enabled etiologic diagnosis in 40–50% of hearing loss patients, but in a large number of cases, a genetic etiology remains inconclusive [2]. In some cases, only a single heterozygous pathogenic or likely pathogenic variant is detected in a gene associated with autosomal recessive hearing loss; in others, the clinical significance of identified variants is uncertain. Relatively common (e.g., allele frequency > 0.3%) silent variants such as synonymous changes that do not alter the amino acid sequence are often removed in the bioinformatic analysis because most are classified as likely benign according to the American College of Medical Genetics and Genomics and the Association for Molecular Pathology (ACMG/AMP) recommendation and the ClinGen Hearing Loss Expert Panel specifications [3, 4].
Autosomal recessive non-syndromic hearing loss (ARNSHL) at the DFNB3 locus on the short arm of chromosome 17 in band p11.2 is caused by biallelic pathogenic variants in MYO15A, a gene of 66 exons that spans ~71 kb and encodes the unconventional myosin XV [5]. Loss of MYO15A function typically leads to congenital profound hearing loss in humans and deafness and vestibular defects in mice [6]. Since DFNB3 was first described [7], more than 200 pathogenic variants in MYO15A have been reported, including some in the Ashkenazi Jewish (AJ) population [8,9,10,11]. Residual hearing have been reported in patients with MYO15A variants, mostly in the N-terminal region; the pathogenicity of variants in other regions found in patients with partial hearing loss has not been rigorously assessed according to the ACMG/ClinGen framework [12,13,14,15].
In this study, we implicate as pathogenic a relatively common synonymous variant c.9861 C > T/p.(Gly3287=) in MYO15A identified in the AJ population, present evidence for its pathogenicity in ARNSHL, and summarize the phenotypic characteristics associated with this variant.
Subjects and methods
Patients
Eighty families with nonsyndromic bilateral sensorineural hearing loss of AJ ancestry were genetically screened at Dor Yeshorim (DY) and referred for follow -up clinical genetic testing when indicated (Supplementary information). Retrospective review of genetic testing results identified other families with relevant variants from those who underwent clinical or research genetic testing at the Children’s Hospital of Philadelphia (CHOP), the Molecular Otolaryngology and Renal Research Laboratories (MORL), the Laboratory for Molecular Medicine (LMM), and Tel Aviv University (TAU). Pedigrees with variants discussed in this study are shown in Fig. 1. Clinical information including newborn hearing screening results, age at onset, and characteristics of hearing loss were retrieved from patients’ medical records, clinical genetic test requisition documents, and direct communications with study participants. Jewish population controls not known to have hearing loss include 9596 anonymous samples (6487 Ashkenazi, 1727 Sephardi, and 1382 both). This study was conducted in compliance with protocols approved by the Institutional Review Boards of DY, Partners HealthCare, the American University of Beirut, the CHOP, and the University of Iowa, and the Helsinki Committees of Tel Aviv University and the Israel Ministry of Health. Written informed consent was obtained from all participants.

Squares denote males and circles denote females. Solid symbols represent individuals affected with bilateral sensorineural hearing loss; clear symbols represent unaffected individuals; dotted symbols represent unaffected heterozygous carriers. Arrowheads point to probands. V, c.9861 C > T/p.(Gly3287=); V1, c.8050 T > C/p.(Tyr2684His); V2, c.8183 G > A/p.(Arg2728His); V3, c.8467 G > A/p.(Asp2823Asn); V4, c.8897_8900dup/p.(Ala2968Profs*33); V5, c.4198 G > A/p.(Val1400Met); V6, c.6292 G > A/p.(Asp2098Asn); V7, c.8090 T > C/p.(Val2697Ala); V8, c.4642 G > A/p.(Ala1548Thr); V9, c.707 A > G/p.(Tyr236Cys); V10, c.10584del/p.(Thr3528Profs*26); +, reference allele; ?, genotype unknown; /, variants in trans; *, phase unknown but assumed in trans because the same variants have not been observed in multiple unrelated families. [V9;V10] indicates that V9 and V10 are in cis. In audiograms, L, left; R, right; yo, years old.
Molecular genetic testing
AJ patients were screened by the DY targeted hearing loss variant panel (Supplementary information). Cases in which the genetic cause for hearing loss could not be determined after the DY screening were referred for clinical next-generation sequencing gene panels, including the MORL OtoSCOPE panel [2], the Sema4 hearing loss panel, or clinical exome sequencing at GeneDx or the Hadassah Medical Center (Table 1). Other hearing loss probands were tested by the HEar-Seq panel at TAU [8, 9], by the OtoSCOPE panel at the MORL [2], by the OtoGenome panel at the LMM, or by the Audiome panel at the CHOP. Clinically significant variants were confirmed by Sanger sequencing. Family members were tested by targeted Sanger sequencing of familial variants.
Variant interpretation
Sequenced variants were described using the Human Genome Variation Society (HGVS) nomenclature (http://varnomen.hgvs.org). NM_016239.4 (NC_000017.10) was used as the cDNA reference sequence. Variants were reviewed and classified according to the ACMG/AMP and ClinGen Hearing Loss Expert Panel guidelines [3, 4]. Variants are listed in ClinVar (Table 2). Population frequencies were estimated based on data from the Genome Aggregation database (http://gnomAD.broadinstitute.org). Population-specific carrier frequencies were determined by screening the variant in the general Jewish population, including 6,487 Ashkenazi, 1,727 Sephardi, and 1,382 mixed Ashkenazi and Sephardi Jewish individuals by DY. Computational predictions were obtained from Varcards (http://varcards.biols.ac.cn/) and Human Splicing Finder (HSF) (http://www.umd.be/HSF3/HSF.shtml). Statistical analysis of the odds ratio, 95% confidence interval, p value, and Z score were calculated using MEDCALC (https://www.medcalc.org/calc/odds_ratio.php).
In vitro splicing analysis
In vitro splicing minigene assays were carried out as described [16,17,18]. Briefly, genomic sequence at chr17:18069512-18069966 (hg19) including exon 61 (161 bp) plus 163 and 131 nucleotides from the 5′ and 3′ flanking sequences, respectively, of MYO15A (NM_016239.4) was PCR amplified from a DNA sample heterozygous for the c.9861 C > T variant using gene-specific primers designed with embedded SalI or SacII restriction enzyme recognition sites. After digestion, PCR fragments were ligated into the pre-constructed pET01 Exontrap vector (MoBiTec, Goettingen, Germany). Sequencing of selected colonies confirmed proper orientation of the cloned fragment and identified both wild-type and variant colonies. Next, the wild-type and variant minigenes were transfected in triplicate into HEK293 cells and total RNA was extracted 36 h post transfection using the Quick-RNA MiniPrep Plus kit (ZYMO Research). cDNA synthesis was performed using RNA SuperScript III Reverse Transcriptase (ThermoFisher Scientific, Waltham, Massachusetts) with a primer specific to the 3′ native exon of the pET01 vector. After PCR amplification, products were visualized on a 1.5% agarose gel, extracted and then Sanger sequenced.
Results
Identification of the c.9861 C > T variant in MYO15A
The probands in DY1 and DY2 (Fig. 1 and Table 1) initially had the OtoSCOPE panel test at the MORL. The c.8050 T > C/p.(Tyr2684His) variant in MYO15A was identified in DY1; DY2 carried the c.8183 G > A/p.(Arg2728His) variant. Further investigation by exome sequencing of the DY1 proband at GeneDx reported MYO15A c.8050 T > C/p.(Tyr2684His) as well as the c.9861 C > T/p.(Gly3287=) variant, which was classified as likely benign at the time because of its relatively high allele frequency (0.4% in AJ) and predicted synonymous impact. The c.9861 C > T/p.(Gly3287=) variant had been filtered out bioinformatically and not reported by the MORL because of its predicted synonymous impact. Follow-up segregation studies confirmed that c.9861 C > T was in trans with c.8050 T > C and co-segregated with hearing loss in DY1 (Fig. 1). Targeted screening of c.9861 C > T and reanalysis of sequencing data confirmed its trans configuration with c.8183 G > A and co-segregation with hearing loss in DY2 (Fig. 1 and Table 1).
Because genetic results from families DY1 and DY2 raised the possibility that c.9861 C > T was potentially pathogenic, the variant was added to the DY hearing loss screening panel. Additional genetically undiagnosed AJ families with hearing loss were screened, and c.9861 C > T was identified in DY3 and DY4 with previously unsolved exome sequencing at Hadassah Medical Center (Table 1). DY5 was positive for the same two variants identified in DY2 and DY4. DY6, DY7, and DY8 were found to have c.9861 C > T and second alleles identified by additional tests (Table 1).
Classification of c.9861 C > T variant in MYO15A according to ACMG/AMP criteria
The c.9861 C > T MYO15A variant has been identified in the heterozygous state in 0.5% (63/12,974) of alleles in the general AJ population and in 4.4% (7/160) AJ alleles in hearing loss probands by DY. It has also been identified in the heterozygous state in 0.4% (42/10,362) of AJ alleles in gnomAD (https://gnomad.broadinstitute.org/variant/17-18069748-C-T). The allele frequency in Sephardi and mixed Ashkenazi and Sephardi Jewish populations are 0.06% (2/3,454) and 0.14% (8/2,764), respectively. This allele is significantly enriched in hearing loss patients over the general AJ population (odds ratio 9.4, 95% confidence interval 4.2–20.8, Z = 5.5, p < 0.0001), thus providing strong evidence for pathogenicity (PS4) [3, 4]. The variant segregates with phenotypes in nine affected and 23 unaffected siblings of the probands in eight AJ families, which provided strong segregation evidence (PP1_Strong) [4]. It was found in one homozygous and 11 compound heterozygous probands with hearing loss (Fig. 1). The variant was in trans with seven different alleles including two variants classified as pathogenic and four as likely pathogenic (Tables 1 and 2). However, because the allele frequency of c.9861 C > T is >0.3% in the AJ population, we considered the allelic evidence moderate (PM3) instead of strong.
The c.9861 C > T variant is predicted to alter splicing by HSF by either activating a cryptic donor site, creating a novel exon splicing silencer (ESS) motif and/or abolishing an exonic splicing enhancer (ESE) motif. To characterize the impact of c.9861 C > T on RNA splicing, we cloned the wild-type and variant sequences of MYO15A (NM_016239.4) exon 61 and flanking introns into the pET01 exon trap vector and transfected them into the HEK293 cell line. Visualization of the splicing products showed that cells transfected with the wild-type vector yielded the expected 407-bp band, which contains exon 61 of MYO15A (Fig. 2). In contrast, cells transfected with the variant construct yielded a single 246-bp band, which corresponds to splicing of native 5′ and 3′ exons of the pET01 vector and skipping of the cloned exon 61 of MYO15A (Fig. 2). Sequencing of purified PCR products confirmed breakpoints and splicing events. No differences were detected among replicates. Skipping of the 161 bp exon is predicted to cause a frameshift that would lead to premature protein truncation. The C > T transition at c.9861 position is computationally predicted to create a cryptic donor site (GT) at c.9860_9861from the reference sequence GC. Should the cryptic donor site be used, c.9860_9948 of 89 bp in exon 61 would be deleted and result in a frameshift that would lead to premature protein truncation. We did not detect the activation of the predicted cryptic donor site as a splice event in our experiments. Results of minigene assays provide functional evidence to support pathogenicity, but the in vitro study may not fully recapitulate the impact under physiological conditions in vivo; therefore, we considered the functional evidence supporting (PS3_Supporting).

a Electrophoresis of RT-PCR products from total RNA extracted from HEK293 cells transfected with wild type, c.9861 C > T, or empty vectors. b Schematic drawings of minigene constructs. Boxes indicate exons. The blue box in the middle is either wild-type (with C) or variant (with T). Spliced products and sequencing results of the RT-PCR products are shown on the right.
In summary, the c.9861 C > T variant in MYO15A is classified as pathogenic based on PP1_Strong, PS4, PM3, and PS3_Supporting according to the ACMG/AMP recommendation for sequence variant interpretation and specifications of the ClinGen Hearing Loss Working Group Expert Panel [3, 4].
Clinical characteristics of patients with c.9861 C > T in MYO15A
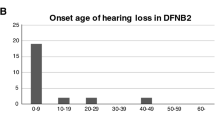
We identified 12 probands and nine affected siblings with biallelic MYO15A variants including c.9861 C > T (Table 1 and Fig. 1). Affected individuals all had bilateral nonsyndromic sensorineural hearing loss, but none had congenital profound deafness typically found in patients with variants that impact MYO15A function [19]. Eight of ten individuals with newborn screening results passed the screening and subsequently developed mild-to-moderate hearing loss, which was typically noticed during early childhood. The loss progressed to moderate-to-severe and even profound with advancing age.
Discussion
Comprehensive genetic testing using high-throughput sequencing generates a large number of genetic variants that require interpretation. Synonymous variants are often excluded from this list as they are typically considered likely benign unless there is compelling evidence for the contrary or they have extremely low allele frequencies. It is known that synonymous variants may be disease-causing by several mechanisms including altered translation rate, mRNA secondary structure, or RNA splicing [20,21,22,23,24,25]. In some instances, multiple mechanisms work together in inducing disease phenotype [23]. However, computational tools to predict these effects are not always reliable [26]. Prior to this study, c.9861 C > T was classified as likely benign because it was found in 0.4% of AJ, which is above the threshold for Criterion BS1, and it is not evolutionarily conserved at the nucleotide level [4]. However, its allele frequency is consistent with the carrier frequency for ARNSHL, meaning that BS1 criterion is not applicable in light of conflicting evidence. ARNSHL is a genetically heterogeneous condition with an estimated prevalence of >1 in 10,000 in various populations, including AJ as well. The prevalence of deafness is not particularly higher in AJ than in other populations. An allele frequency of 0.4% would be consistent with carrier frequency for ARNSHL in any population. The difference among different populations are different major contributors that need to be included on the exception list of ClinGen allele frequency cutoff rules. Our data established the pathogenicity of the variant based on strong segregation, statistical, moderate allelic, and supporting functional evidence to indicate that abnormal splicing is a probable cause (PP1_Strong, PS4, PM3, PS3_Supporting) [3, 4].
Establishing the pathogenicity of c.9861 C > T, together with other specific variant criteria, allowed us to classify c.8090 T > C/p.(Val2697Ala) as pathogenic, which led to classification of c.4642 G > A/p.(Ala1548Thr) as pathogenic. Hence, conclusive diagnoses were reached for 13 families: DY1, DY2, DY3, DY4, DY5, DY6, DY7, DY8, TAU9, MORL10, LMM12, LMM13, and CHOP14. Additional data are required to clarify the clinical significance of c.6292 G > A/p.(Asp2098Asn) identified in LMM11.
Patients with c.9861 C > T reported in this study presented with delayed-onset, mild-to-moderate hearing loss, as opposed to the congenital profound deafness typically observed in individuals with MYO15A-related hearing loss, suggesting the splicing defect of c.9861 C > T may be leaky. Notably, the majority of patients with c.9861 C > T passed newborn hearing screening, developed hearing loss during early childhood, and had a progressive loss when long-term follow -up information was available. Because this variant is relatively common in the AJ population, genetic screening of this variant may identify carrier parents and newborns with AJ ancestry at risk of hearing loss.
In conclusion, the c.9861 C > T variant in MYO15A is a relatively common synonymous variant enriched in the AJ population. It leads to abnormal splicing. Individuals homozygous or compound heterozygous for the variant show childhood onset progressive hearing loss.
Data availability
The variants are available on ClinVar (Accession numbers): c.9861 C > T/p.(Gly3287=) (SCV000803286.1), c.8050 T > C/p.(Tyr2684His) (SUB8394617), c.8183 G > A/p.(Arg2728His) (SUB8394617), c.8090 T > C/p.(Val2697Ala) (SCV0000626 66.6), c.4642 G > A/p.(Ala1548Thr) (SCV000272098.2), c.6292 G > A/p.(Asp2098Asn) (SCV000199408.4).
References
Morton CC, Nance WE. Newborn hearing screening-a silent revolution. N Engl J Med. 2006;354:2151–64.
Sloan-Heggen CM, Bierer AO, Shearer AE, Kolbe DL, Nishimura CJ, Frees KL, et al. Comprehensive genetic testing in the clinical evaluation of 1119 patients with hearing loss. Hum Genet. 2016;135:441–50.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24.
Oza AM, DiStefano MT, Hemphill SE, Cushman BJ, Grant AR, Siegert RK, et al. Expert specification of the ACMG/AMP variant interpretation guidelines for genetic hearing loss. Hum Mutat. 2018;39:1593–613.
Wang A, Liang Y, Fridell RA, Probst FJ, Wilcox ER, Touchman JW, et al. Association of unconventional myosin MYO15 mutations with human nonsyndromic deafness DFNB3. Science. 1998;280:1447–51.
Liang Y, Wang A, Belyantseva IA, Anderson DW, Probst FJ, Barber TD, et al. Characterization of the human and mouse unconventional myosin XV genes responsible for hereditary deafness DFNB3 and shaker 2. Genomics 1999;61:243–58.
Friedman TB, Liang Y, Weber JL, Hinnant JT, Barber TD, Winata S, et al. A gene for congenital, recessive deafness DFNB3 maps to the pericentromeric region of chromosome 17. Nat Genet. 1995;9:86–91.
Brownstein Z, Friedman LM, Shahin H, Oron-Karni V, Kol N, Abu Rayyan A, et al. Targeted genomic capture and massively parallel sequencing to identify genes for hereditary hearing loss in Middle Eastern families. Genome Biol. 2011;12:R89.
Brownstein Z, Abu-Rayyan A, Karfunkel-Doron D, Sirigu S, Davidov B, Shohat M, et al. Novel myosin mutations for hereditary hearing loss revealed by targeted genomic capture and massively parallel sequencing. Eur J Hum Genet. 2014;22:768–75.
Azaiez H, Booth KT, Ephraim SS, Crone B, Black-Ziegelbein EA, Marini RJ, et al. Genomic landscape and mutational signatures of deafness-associated genes. Am J Hum Genet. 2018;103:484–97.
Nal N, Ahmed ZM, Erkal E, Alper OM, Luleci G, Dinc O, et al. Mutational spectrum of MYO15A: the large N-terminal extension of myosin XVA is required for hearing. Hum Mutat. 2007;28:1014–9.
Bashir R, Fatima A, Naz S. Prioritized sequencing of the second exon of MYO15A reveals a new mutation segregating in a Pakistani family with moderate to severe hearing loss. Eur J Med Genet. 2012;55:99–102.
Chang MY, Lee C, Han JH, Kim MY, Park HR, Kim N, et al. Expansion of phenotypic spectrum of MYO15A pathogenic variants to include postlingual onset of progressive partial deafness. BMC Med Genet. 2018;19:29.
Cengiz FB, Duman D, Sirmaci A, Tokgoz-Yilmaz S, Erbek S, Ozturkmen-Akay H, et al. Recurrent and private MYO15A mutations are associated with deafness in the Turkish population. Genet Test Mol Biomark. 2010;14:543–50.
Chang MY, Kim AR, Kim NK, Lee C, Lee KY, Jeon WS, et al. Identification and clinical implications of novel MYO15A mutations in a non-consanguineous Korean family by targeted exome sequencing. Mol Cells. 2015;38:781–8.
Booth KT, Azaiez H, Kahrizi K, Wang D, Zhang Y, Frees K, et al. Exonic mutations and exon skipping: Lessons learned from DFNA5. Hum Mutat. 2018;39:433–40.
Booth KT, Askew JW, Talebizadeh Z, Huygen PLM, Eudy J, Kenyon J, et al. Splice-altering variant in COL11A1 as a cause of nonsyndromic hearing loss DFNA37. Genet Med. 2018;21:948–54.
Booth KT, Kahrizi K, Najmabadi H, Azaiez H, Smith RJ. Old gene, new phenotype: splice-altering variants in CEACAM16 cause recessive non-syndromic hearing impairment. J Med Genet. 2018;55:555–60.
Friedman TB, Liang Y, Weber JL, Hinnant JT, Barber TD, Winata S, et al. A gene for congenital, recessive deafness DFNB3 maps to the pericentromeric region of chromosome 17. Nat Genet. 1995;9:86–91.
Bali V, Bebok Z. Decoding mechanisms by which silent codon changes influence protein biogenesis and function. Int J Biochem Cell Biol. 2015;64:58–74.
Hunt RC, Simhadri VL, Iandoli M, Sauna ZE, Kimchi-Sarfaty C. Exposing synonymous mutations. Trends Genet. 2014;30:308–21.
Kirchner S, Cai Z, Rauscher R, Kastelic N, Anding M, Czech A, et al. Alteration of protein function by a silent polymorphism linked to tRNA abundance. PLoS Biol. 2017;15:e2000779.
Katneni UK, Liss A, Holcomb D, Katagiri NH, Hunt R, Bar H, et al. Splicing dysregulation contributes to the pathogenicity of several F9 exonic point variants. Mol Genet Genom Med. 2019;7:e840.
Nackley AG, Shabalina SA, Tchivileva IE, Satterfield K, Korchynskyi O, Makarov SS, et al. Human catechol-O-methyltransferase haplotypes modulate protein expression by altering mRNA secondary structure. Science. 2006;314:1930–3.
Bartoszewski RA, Jablonsky M, Bartoszewska S, Stevenson L, Dai Q, Kappes J, et al. A synonymous single nucleotide polymorphism in DeltaF508 CFTR alters the secondary structure of the mRNA and the expression of the mutant protein. J Biol Chem. 2010;285:28741–8.
Sauna ZE, Kimchi-Sarfaty C. Understanding the contribution of synonymous mutations to human disease. Nat Rev Genet. 2011;12:683–91.
Brownstein Z, Gulsuner S, Walsh T, Martins FTA, Taiber S, Isakov O, et al. Spectrum of genes for inherited hearing loss in the Israeli Jewish population, including the novel human deafness gene ATOH1. Clin Genet. 2020;98:353–64.
Cengiz FB, Duman D, Sirmaci A, Tokgöz-Yilmaz S, Erbek S, Oztürkmen-Akay H, et al. Recurrent and private MYO15A mutations are associated with deafness in the Turkish population. Genet Test Mol Biomark. 2010;14:543–55.
Manzoli GN, Bademci G, Acosta AX, Felix TM, Cengiz FB, Foster J II, et al. Targeted resequencing of deafness genes reveals a founder MYO15A variant in northeastern Brazil. Ann Hum Genet. 2016;80:327–31.
Cabanillas R, Dineiro M, Cifuentes GA, Castillo D, Pruneda PC, Álvarez R, et al. Comprehensive genomic diagnosis of non-syndromic and syndromic hereditary hearing loss in Spanish patients. BMC Med Genomics. 2018;11:58.
Schrauwen I, Sommen M, Corneveaux JJ, Reiman RA, Hackett NJ, Claes C, et al. A sensitive and specific diagnostic test for hearing loss using a microdroplet PCR-based approach and next generation sequencing. Am J Med Genet Pt A. 2013;161:145–52.
Brownstein Z, Friedman LM, Shahin H, Oron-Karni V, Kol N, Abu Rayyan A, et al. Targeted genomic capture and massively parallel sequencing to identify genes for hereditary hearing loss in Middle Eastern families. Genome Biol. 2011;12:R89.
Yang T, Wei X, Chai Y, Li L, Wu H. Genetic etiology study of the non-syndromic deafness in Chinese Hans by targeted next-generation sequencing. Orphanet J Rare Dis. 2013;8:85.
Brownstein Z, Abu-Rayyan A, Karfunkel-Doron D, Sirigu S, Bella Davidov B, Shohat M, et al. Novel myosin mutations for hereditary hearing loss revealed by targeted genomic capture and massively parallel sequencing. Eur J Hum Genet. 2014;22:768–75.
Fattahi Z, Shearer AE, Babanejad M, Bazazzadegan N, Almadani SN, Nikzat N, et al. Screening for MYO15Agene mutations in autosomal recessive nonsyndromic, GJB2 negative Iranian deaf population. Am J Med Genet Pt A. 2012;158:1857–64.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24.
Oza AM, DiStefano MT, Hemphill SE, Cushman BJ, Grant AR, Siegert RK, et al. Expert specification of the ACMG/AMP variant interpretation guidelines for genetic hearing loss. Hum Mutat. 2018;39:1593–613.
Acknowledgements
We thank patients and family members who participated in the study. We thank Dr. Ian Krantz of Children’s Hospital of Philadelphia for providing phenotypic information on a proband reported in this manuscript, Esther Zlotnik of Dor Yeshorim Israel for collecting patient information and Liliya Moshkovsky of Dor Yeshorim Israel for performing the laboratory work. We dedicate this manuscript in memory of Prof. Maria Bitner-Glindzicz.
Funding
This work was supported by National Institutes of Health / National Institute on Deafness and other Communication Disorder grants R01DC015052 (CCM and JS), R01DC011835 (KBA), R01DC003544, R01DC002842 and R01DC012049 (RJHS), R03DC013866 (JS), and NIHR Manchester Biomedical Research Center (CCM).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
KTB, AMO, KL, SSA, HLR, DK, KF, CN, ML, CF, CCM, RJHS, and JS worked for pay-for-service diagnostic laboratories providing genetic testing. All other authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Hirsch, Y., Tangshewinsirikul, C., Booth, K.T. et al. A synonymous variant in MYO15A enriched in the Ashkenazi Jewish population causes autosomal recessive hearing loss due to abnormal splicing. Eur J Hum Genet 29, 988–997 (2021). https://doi.org/10.1038/s41431-020-00790-w
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-020-00790-w
This article is cited by
-
Using multi-scale genomics to associate poorly annotated genes with rare diseases
Genome Medicine (2024)
-
Genetic etiology of hearing loss in Iran
Human Genetics (2022)
-
Comprehensive molecular-genetic analysis of mid-frequency sensorineural hearing loss
Scientific Reports (2021)
