
Abstract
Participation of clinical genetic laboratories in External Quality Assessment schemes (EQAs) is a powerful method to ascertain if any improvement or additional training is required in the diagnostic service. Here, we provide evidence from recent EQAs that the competence in recognizing and interpreting cytogenetic aberrations is variable and could impact patient management. We identify several trends that could affect cytogenomic competence. Firstly, as a result of the age distribution among clinical laboratory geneticists (CLGs) registered at the European Board of Medical Genetics, about 25–30% of those with experience in cytogenetics will retire during the next decade. At the same time, there are about twice as many molecular geneticists to cytogeneticists among the younger CLGs. Secondly, when surveying training programs for CLG, we observed that not all programs guarantee that candidates gather sufficient experience in clinical cytogenomics. Thirdly, we acknowledge that whole genome sequencing (WGS) has a great attraction to biomedical scientists that wish to enter a training program for CLG. This, with a larger number of positions available, makes a choice for specialization in molecular genetics logical. However, current WGS technology cannot provide a diagnosis in all cases. Understanding the etiology of chromosomal rearrangements is essential for appropriate follow-up and for ascertaining recurrence risks. We define the minimal knowledge a CLG should have about cytogenomics in a world dominated by WGS, and discuss how laboratory directors and boards of professional organizations in clinical genetics can uphold cytogenomic competence by providing adequate CLG training programs and attracting sufficient numbers of trainees.
Similar content being viewed by others
Introduction
With the prospect of high diagnostic yields and decreasing costs per sample, whole genome sequencing (WGS) is emerging as a first-tier test for many referrals to diagnostic genome laboratories. WGS enables the detection of pathogenic gene variants, copy number gains and losses, and loss of heterozygosity in a single genetic test [1,2,3]. In addition, about 90% of breakpoints of balanced rearrangements can be identified using WGS [4]. In Supplementary Box 1 and Supplementary Tables 1–3 we describe the huge potential of WGS in genome diagnostics. The upcoming transition to WGS will have immense benefits to the patients and their families as more patients will receive a diagnosis. In Supplementary Table 1 we show that WGS has a much higher diagnostic yield than karyotyping in the traditional postnatal referral categories. For this reason, we expect that WGS will supplant karyotyping and chromosome microarray (CMA) investigation as a standard, first-tier genetic test within the next decade. Because of the associated cost per sample, this transition will take place initially in high-income countries (HICs, as defined by the World Bank). In low- and middle-income countries (LMICs) karyotyping will remain a prominent genetic testing method, as explained in Supplementary Box 2 and Supplementary Table 4.
Despite a transition to WGS, there will be a continuous need for clinical laboratory geneticists (CLGs) that are competent in cytogenetics. The reasons for this are twofold. Firstly, using short-read, paired-end sequencing by synthesis technology at 30x average genome coverage, the current standard for WGS, a significant proportion of genome aberrations of clinical significance cannot be detected, such as low-level mosaicism, Robertsonian translocations, small supernumerary marker chromosomes (sSMC) and balanced rearrangements with breakpoints that are not in unique DNA sequences. Together these represent about 8% of the current referrals for postnatal karyotyping [5]. Secondly, guidelines for cytogenetic analysis state that imbalances detected by CMA should receive appropriate follow-up studies by karotyping and/or fluorescence in situ hybridization (FISH) in order to determine the structural rearrangement underlying the imbalance [6, 7]. The same would apply when an imbalance is detected by WGS and the data are inconclusive with respect to the underlying chromosomal rearrangement. An example is shown in Fig. 1. It is important for genetic counseling and for the determination of the recurrence risk to precisely identify the type of structural rearrangement.

Current WGS methods based on short-read, paired-end sequencing are not suited to discriminate between these possible rearrangements because breakpoints that are located within repetitive DNA sequences are not recognized [4]. In contrast, these rearrangements can be discriminated by microscopy, using karyotyping and/or FISH. The simplest structural rearrangement is a terminal deletion although an interstitial deletion, a ring chromosome, or a derivative chromosome representing an unbalanced translocation involving chromosome 15 and the very terminal end of another chromosome arm (here 20p) can underlie such a terminal loss. As emphasized in current guidelines for cytogenomic investigation [7], identification of the underlying rearrangement is essential for determining the recurrence risk for the parents of the patient, for genetic counseling and for the identification of family members who are at an increased risk of having imbalanced progeny.
To ensure the competence of CLGs, professional registries have been established, for example, in the United States by the American College of Medical Genetics and Genomics (ACMMG) [8], and in Europe by the national societies of human genetics and by the European Board of Medical Genetics (EBMG) [9], and by state registration in some countries. Accredited training programs for CLGs have been established as well [10] and the duties, tasks, and responsibilities of CLGs have been defined [11]. In most countries it is the task of the CLG to produce the reports of the laboratory findings to the referring clinicians [11]. Competence is also ensured by internationally accepted standards and guidelines for the analysis and interpretation of cytogenetic results [7, 12]. An important method for assessing the competence of CLGs is by regular participation in External Quality Assessment (EQA) schemes, as is required for diagnostic laboratories when adopting the international standard ISO 15189 [13]. EQA can examine the analytical and interpretative skills of laboratories and CLGs against international guidelines under conditions that mimic reality as closely as possible. Following an initial observation from an EQA in postnatal karyotyping on fading analytical competence [14], we here present novel observations from multiple, more recent EQAs that indicate that the competence of clinical genome laboratories is under constraint, both at the analytical and interpretative levels, and not only for light microscopy but also for molecular genetic methods. We describe the results of these EQAs and discuss the threats that affect both cytogenetic competence and molecular analysis of aneuploidies as well as possible interventions that could be made to preserve cytogenomic competence for future generations.
We define “cytogenomics” (a merger of “cytogenetics” and “genomics”) as the study of the numerical and structural variation of the genome at the chromosomal and subchromosomal level using methods that cover the entire genome or specific DNA sequences. These include, for example, light microscopy (karyotyping), molecular cytogenetic methods (FISH), and molecular genetic methods such as quantitative fluorescent polymerase chain reaction (QF-PCR), CMA, optical mapping, and WGS.
Recent EQA schemes show that fading cytogenomic competence is widespread
A survey of recent EQAs provided by GenQA shows that the ability of laboratories for clinical genetics to recognize cytogenomic abnormalities and reporting them to the referring clinician is variable, both in constitutional and acquired abnormalities. The cases chosen for these EQAs were realistic and were representative for the workload of most laboratories. In addition, all cases were independently assessed blind (i.e., without knowing the result) by multiple assessors, and considered not to be excessively difficult or complex before being made available to the participating laboratories. Therefore, we believe that these EQA cases did not exceed the expected level of competence. As a consequence, a failure to meet the internationally accepted standards for analyzing and interpreting cytogenomic aberrations in these cases is indicative of insufficient training and/or basic knowledge. Several recurrent errors were observed at both the analytical and interpretative levels in these EQAs. We chose only to discuss EQAs where a minimum of five participants made a similar, grave error with consequences for clinical management of the patient. These are summarized in Table 1. For comparison, we included for each EQA the base line percentage of such grave errors, based on the most recent 5 years.
When assessing analytical skills, it appeared that many participants failed to recognize an abnormal chromosome in karyograms made from G-banded metaphase images. For example, 29.3% of participants failed to recognize an idic(Y)(q11.?12) in a male with infertility in G-banded metaphases from peripheral blood lymphocytes [14]. In a bone marrow sample from a patient with acute myeloid leukaemia (AML), 10.8% of participants did not recognize a translocation t(9;11)(p21;q23) in G-banded metaphases, and 6.3% did not recognize this rearrangement by FISH using a KTM2A break apart probe. Such recurrent chromosome rearrangements in AML [15] should be known to CLGs working in the field of haematological disorders. Whereas molecular testing of the infertile male would have identified a loss of the AZF fertility genes on the long arm of the Y-chromosome as the cause of infertility, and FISH with a KMT2A probe would have identified the translocation t(9;11)(p21;q23) in the AML-patient, these two EQA examples imply that if karyotyping had been the only test in these cases, these structural chromosome abnormalities would not have been identified. A further lack of cytogenomic analytical skills became apparent when 19% of participating laboratories did not recognize trisomy 13 mosaicism in a short tandem repeat (STR) marker pattern obtained by QF-PCR on DNA from chorionic villi sampling (CVS).
When assessing interpretative skills, specific knowledge gaps were highlighted through the results of these EQAs (Table 1). These include a failure by 3.5% of participants to recognize that the carrier of a balanced translocation can transmit the smallest derivative chromosome as a supernumerary chromosome to the child as the result of a 3:1 meiotic segregation. This demonstrates an insufficient knowledge about the mechanisms by which balanced autosomal reciprocal translocations can lead to genomic imbalances in the progeny, as described, for example, in a widely used textbook [16]. A gap in fundamental knowledge about the embryological origin of the fetus and the placenta [16] is illustrated by the failure of 17% of participants to recognize the possibility of confined placental mosaicism in a case of trisomy 13 mosaicism detected by QF-PCR in chorionic villi from a pregnancy without ultrasound abnormalities.
We conclude that the decrease in analytical and interpretative skills of cytogenomic anomalies is widespread among laboratories that offer cytogenetic and QF-PCR based genetic services. Firstly, in three EQAs more than 10% of participating laboratories had a poor performance (range 10.8–29.3%). For each of these EQAs, this exceeds by far the expected base line percentage of significant errors resulting in poor performance (Table 1). Secondly, there is little correlation between the participants with poor performance in the different EQAs as, for example, only three of the 48 poor performance designations in the 2016 Postnatal Blood EQA were also included in the 2018 Prenatal Molecular Rapid Aneuploidy Screening EQA. Thirdly, there is no evidence that the poor performance is higher among laboratories participating in an EQA for the first time (in the EQA with the highest percentage of poor performance designations, only 9% were first-time participants). Fourthly, the recurrent poor performances covered the full spectrum of diagnostic cytogenomic investigation, both with respect to the methods of investigation (including both light microscopy and molecular methods) and the type of referrals (including post- and prenatal constitutional and acquired abnormalities).
Clinical consequences for patient management of fading cytogenomic-based expertise
The errors seen in the EQAs described in Table 1 have adverse consequences for patient management. This is most dramatically seen in the acute leukemia patient where 10.8% of laboratories missed a recurrent translocation t(9;11)(p21;q23), which has an intermediate prognosis [15]. In a diagnostic setting this would have resulted in the referring hematologist receiving incorrect information resulting in inappropriate patient management. The inability to correctly associate an observed imbalance in the progeny to the corresponding balanced rearrangement in the carrier parent reflects a lack of knowledge about the meiotic segregation patterns of balanced chromosomal rearrangements. Not knowing or understanding the meiotic origin of imbalances will provide incorrect or incomplete information to the referring physician about the recurrence risk in future pregnancies.
In prenatal diagnosis, there is also a potential for very severe, adverse consequences for the patient, as shown by the EQA for rapid prenatal aneuploidy testing using QF-PCR based on DNA from chorionic villi. Here, 19% of participating laboratories failed to recognize that the CVS sample was mosaic for trisomy-13 and 17% failed to recognize that the trisomy-13 could be confined to the placenta (Table 1). Such erroneous interpretations could lead to termination of a non-trisomic pregnancy if appropriate follow-up by amniocenteses was not recommended. This EQA example shows that a large proportion of laboratories that offer this test demonstrate an inadequate knowledge of the biology of the placenta and of the origin of chromosomal mosaicism during early pregnancy. This is a matter of concern because such basic knowledge is equally essential to correctly interpret and manage the additional findings associated with the novel NIPT test (noninvasive prenatal testing). NIPT is based on cell-free fetal DNA in the maternal circulation that has its origin in apoptotic cytotrophoblasts of chorionic villi that are derived from the developing placenta. Consequently, NIPT is not a diagnostic test of the fetus proper. Known causes of discordant results between NIPT and the true fetal karyotype include confined placental mosaicism, maternal malignancy, a vanishing twin, maternal copy number variation, a low level of maternal mosaic aneuploidy and true fetal mosaicism. These lead to a false positive result of trisomy 13, 18 or 21 in about 88% of discordant cases and a false negative result in about 12% [17]. Whereas NIPT was initially designed to detect trisomy 13, 18, and 21, a large proportion of women may opt for genome-wide analysis (currently this is 78% in The Netherlands [18]). Additional findings (i.e. abnormal findings other than trisomy 13, 18, 21, many of which are confined to the placenta), occur in about 1 in 300 cases in a low-risk population [18]. Discordant findings and additional findings must be recognized and adequately reported to the referring gynecologist in order to instigate appropriate follow-up studies to ascertain whether the fetus itself is affected or not. Likewise, positive NIPT results of a trisomy 13 and 21 must be followed by karyotyping to discriminate a free trisomy from an unbalanced Robertsonian translocation in order to determine the recurrence risk.
Factors affecting the competence in cytogenomics now and in the next decade
In most laboratories, CLGs are the professionals who are responsible for interpreting the laboratory results in the context of the referral reason and for writing the laboratory report [11]. Thus, a laboratory must have a sufficient number of CLGs that are competent to fulfil this task, and therefore, we have to focus on these professionals to identify the factors that contribute to the observed fading of cytogenomic competence. According to ISO 15189, the laboratory must identify the root cause of a significant error that affects patient management and it must take appropriate measures to prevent this error from happening again [19]. As a root cause analysis is an intra laboratory process we cannot know the exact reasons for the observed errors. Nevertheless, it is possible to identify some general trends that will affect the number of CLGs that are sufficiently competent during the next decade. As a consequence, these trends will affect the competence of diagnostic laboratories and may place the clinical management of patients at risk. Although medical doctors (MDs) with a specialization in clinical genetics also sign reports in some countries [11], here we focus on the people doing the practical work, which in most cases are the CLGs. We identified the following problems.
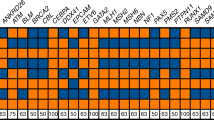
Firstly, the history and demography of the field of medical genetics imply that a large proportion of CLGs who specialized in clinical cytogenetics at the beginning of their career will retire during the next decade. During the nineteen seventies, easy-to-use and cheap methods for culturing human T-lymphocytes and for obtaining banded metaphase chromosomes were developed [20]. This attracted many young biologists with an interest in genetics to the rapidly developing field of clinical cytogenetics. Most of the cytogeneticists who entered the field during that time were around 25–30 years of age and, consequently, most of them have retired by now. The age distribution of CLGs from the EBMG Registry shows that during the next decade 25–30% of currently active CLGs who specialized in clinical cytogenetics will also retire (Fig. 2). Many of these CLGs have extensive experience and their expertise will be lost. In comparison, only 15–20% of the EBMG-registered molecular geneticists will retire in the next ten years. A shortage of cytogenetic CLGs may already occur in some countries and it is known that retired staff have been asked to assist in public and private diagnostic genome laboratories (personal communication of authors).

Age groups are in years. Clinical cytogeneticists and biochemical geneticists (others) are overrepresented in the older age groups compared to molecular geneticists (molgen).
Secondly, young CLGs have a preference for molecular genetics. There are no solid empirical data to support this statement but when looking at the EBMG-registered CLGs, molecular geneticists are overrepresented among the younger age groups. For example, in the youngest age group there are twice as many molecular geneticists compared to the other specializations (Fig. 2). This may also be true for those entering the field. One of the underlying factors may be the anticipated growth and benefits of WGS (as detailed in Supplementary Box 1), which also have an inherent appeal to the general public. This can be illustrated by the life-saving effects of rapid WGS on the clinical management of critically ill babies within the dramatic setting of the neonatal intensive care unit [21,22,23]. In addition, the rapid technical innovations and anticipated cost reductions in DNA sequencing technology, together with advertising campaigns by biotechnology and sequencing companies will further lead to a growth of the number of referrals and will also add to the attractiveness of WGS to young biologists. And finally, it is natural for young medical biologists, after having held temporary appointments as Ph.D. student or postdoctoral researcher, to choose a career in a field where there is ample opportunity for obtaining permanent jobs in academia, government and industry. For example, for the USA, the indeed.com website listed about 220 positions in the category “Clinical Cytogenetics” and 2312 in the category “Clinical Molecular Genetics” [24], a 10-fold difference. Also, the annual growth rate of the economic value of the global next-generation sequencing (NGS) market is predicted at about 12% until 2027, with clinical diagnostic laboratories accounting for the largest share in this market [25]. All these factors may influence the ambitions and career decisions of those that enter the CLG training programs in such a way that they opt for molecular genetics, not for cytogenomics.
Thirdly, we observe that not all training programs for CLGs accommodate a specified minimum time period for cytogenomics and a specified minimum number of cases that must be solved by the trainee. This is based on a survey of training programs from representative countries (Table 2). Because of the rapid technological developments in the field we only included programs that had recently been updated. In total, 16 programs were selected. The duration of a full-time training program varies from two years (Canada, Sweden) to five years (Austria, Australia, Germany, New Zealand). The majority of the training programs (11 of 16) have a duration of at least four years. Three countries (Canada, Norway, Sweden) have separate programs for cytogenomics and molecular genetics, whereas 13 out of 16 countries have a combined program for both. In seven of these combined programs there is no specification of the duration of the period for dedicated training in cytogenomics. In 11 of these programs the minimal number of microscopic analyses is not specified, and in eight programs the minimal number of cytogenomic reports is not specified (Table 2). While we do not wish to comment on the contents and organization of these training programs, it is a matter of concern that the majority of the programs did not contain a specified minimum workload of cytogenomic cases that must be successfully solved by the trainee, as the diversity of chromosome abnormalities and their associated complex consequences for patient management [16] require a prolonged immersion in the environment of a diagnostic cytogenetics laboratory. Trainees need to ensure that they will be able to adequately analyze, interpret and report realistic and representative cases as described in Table 1. We are concerned that the further growth and successes of WGS (see Supplementary Box 1) may lead to reductions of the period dedicated to training in cytogenomics, as has occurred in The Netherlands [14]. Likewise, in countries where MDs need to sign laboratory reports [11], the education of MDs must include knowledge about clinical cytogenomics. However, in the German training program for MDs becoming specialists in clinical genetics the time for education in cytogenetics was recently reduced from 12 to 6 months [26]. Whereas a reduction in microscopy is justifiable given the implementation of molecular methods with higher resolution, the training programs must ensure that the basic knowledge about the mechanisms that cause pathogenic chromosomal aberrations is retained.
In conclusion, if not appropriately addressed, the field of diagnostic genetics will face a deficit in CLGs (and MDs) that are competent in cytogenomics in the next decade. This is due to retirement of 25–30% of cytogeneticists, a preference of young trainees for molecular genetics, an excess of job opportunities in molecular genetics and a tendency to accommodate less time for cytogenomics in many training programs, all occurring more or less simultaneously (as depicted in Fig. 3). At the same time, an anticipated reduction in the number of referrals for light microscopy will reinforce some of these factors. For example, a reduction in the number of referrals for light microscopy will affect cytogenomic competence in the long-term because it may influence those in charge of training programs to accommodate less time for cytogenomics and may influence young trainees not to choose this specialization.

Please note that this diagram is only applicable for countries with sufficiently large populations and a financially well-equipped health care system.
Responsibilities and recommendations to preserve cytogenomic competence
The preservation of the quality of a diagnostic cytogenetic service is, above all, a responsibility of each individual laboratory. Laboratory directors should ensure that the laboratory staff contains a sufficient number of competent CLGs. If appropriate, the laboratory should anticipate the scheduled retirement of experienced cytogeneticists by attracting sufficient numbers of trainees. In addition, on a national level, the organizations responsible for the training of CLGs should provide trainees with a program that corresponds to the changing diagnostic methods and, at the same time, assures that their competence in the analysis, interpretation, and reporting of cytogenomic referrals is at an adequate level. Finally, at an international level, this is also true for the boards of professional organizations such as the American College of Medical Genetics and Genomics (ACMGG), the European Society of Human Genetics (ESHG), the EBMG, and the European Cytogeneticists Association (ECA), who have a shared responsibility in providing regular updates of quality guidelines and professional training programs in this rapidly changing field.
Given the multiple benefits of WGS (Supplementary Box 1), actions that are effective on the long term should be focused on attracting talented and motivated trainees and on providing them with an attractive and adequate training program in clinical cytogenomics. In order to do this it is necessary to carefully define the minimum knowledge a CLG specializing in cytogenetics should have ten years from now, when WGS will be the first-tier genetic test in the vast majority of referrals to diagnostic genome laboratories in HICs with a well-developed health care system. We propose that, firstly, CLGs will be needed who are able to recognize the signature of chromosomal aberrations in WGS data in order to instigate the appropriate follow-up studies by microscopy, with or without FISH. Thus, we may need a different kind of CLG, who is able to analyze WGS data from a cytogenomic perspective. Such CLGs are also required to instigate and interpret an analysis by light microscopy when the WGS data are not fully informative with respect to the structure of an observed genomic imbalance. In addition, when losses or gains are identified by WGS, it is essential that the etiology of the different types of rearrangements are understood when parental analysis is undertaken to identify or exclude an associated balanced chromosomal rearrangement [6]. Future CLGs must be able to recognize alterations in chromosome banding patterns when suspected rearrangements are detected using molecular methods, such as WGS and the more recently established long-range sequencing [27] and optical mapping methods [28,29,30]. We consider that training of cytogenetic CLGs in the analysis of CMA results can bridge the gap between G-banding and WGS, because these CLGs are able to recognize the mechanisms that produce the copy number changes revealed by CMA, such as the unbalanced segregation products of a balanced rearrangement, sSMCs, mosaicism and structural aberrations of the sex chromosomes. Because CNVs can also be detected in sequencing data, it will be these CLGs who, during the next decade, are able to provide adequate training in cytogenomics to trainees entering the field in the WGS era.
Secondly, we propose that we need CLGs who are aware of the frequencies and types of chromosomal aberrations of clinical relevance that are not detectable by WGS technology and that, for this reason, cannot be excluded as the cause of the clinical symptoms of the patient (e.g., ring(20) epilepsy syndrome, low-level mosaicism, rearrangement with heterochromatic breakpoints) [4, 5, 31, 32]. A survey during a ten-year period at a single center shows that such cases undetectable by current WGS technology comprise about 8% of all referrals for postnatal karyotyping [5]. Thus, the cytogenomic specialist should know when a balanced parental rearrangement of clinical significance may be involved that is not revealed by WGS. For gains and losses detected by WGS, the CLG should know whether chromosome analysis or FISH on metaphases is most appropriate. With respect to sSMCs, the CLG should be aware that these may be mitotically unstable and, therefore, frequently occur in a fraction of the cells [31, 32], possibly below the detection limit of current WGS technology [33, 34]. Also when other types of chromosomal mosaicism are suspected, as indicated by clinical symptoms such as body asymmetry and skin pigmentation patterns [35], the cytogenomic expert should apply FISH of uncultured cells, as this can reveal low-level mosaicism of clinical relevance that is not detectable by WGS [36].
Thirdly, we propose that CLGs need to be aware when mosaicism detected in postnatal and prenatal samples is not related to the clinical referral reason. For example, in postnatal samples, age-related sex chromosome loss of the X chromosome in females and Y chromosome in males must not be confused with a 45,X/46,XX karyotype seen in younger females with Turner syndrome, or Y chromosome loss in hematological samples [16]. In prenatal samples, mosaicism in chorionic villus or NIPT samples can be confined to the placenta [37].
As karyotyping will remain an affordable method in many LMICs, being the only genetic test available to the majority of the world’s children with serious birth defects (Supplementary Box 2), there is an additional need to preserve cytogenomic competence in countries that cannot afford the transition to clinical WGS in the next decade. Thus, CLGs that perform karyotyping in these countries must remain competent to identify subtle alterations of banding patterns of metaphase chromosomes. In addition, as NIPT is being implemented in public health care in many LMICs [38,39,40], the cytogenetic follow-up of positive and additional findings, as discussed above, will also add to the workload of CLGs performing karyotyping in these countries.
Concluding remarks: a future for clinical cytogenomics in the WGS era
The cytogenomic specialist of the future must be capable of recognizing the need for follow-up studies by light microscopy if the WGS data are indicative of a chromosomal rearrangement. The CLG must be aware of the limitations of WGS technology, and, if appropriate, will apply other methods of genetic investigation to obtain all the information needed for adequate clinical management of the patient. Thus, there will be a continued need for experts in clinical cytogenomics during the next decade. There are ample possibilities to promote the field of clinical cytogenomics and attract sufficient capable trainees. Firstly, it should be made clear to trainees entering the field that the work of a CLG specializing in cytogenomics will become more diverse and more interesting because new methods and technologies are emerging that lead to the identification of novel mechanisms of genetic disease (summarized in Table 3). For example, about 7% of de novo balanced rearrangements in MCA/MR patients lead to the disruption of TAD organization, providing a plausible explanation for the clinical phenotype [4]. During the coming decade, these novel, genome-wide approaches will make clinical cytogenomics more attractive and more challenging as they will be validated for use in clinical diagnostics. Secondly, the training programs for CLGs could be made more attractive by accommodating time for trainees to immerse themselves in these novel technologies and by stimulating exchanges of trainees, both nationally and internationally, between laboratories that are validating these novel methods for clinical use. Thirdly, it should be explained to trainees that the universe of pathogenic variants in Mendelian disorders is finite and that, therefore, the reporting of both normal and pathogenic findings in the clinical molecular investigation of monogenic diseases will be dominated by automated systems that require limited human intervention (as explained in Supplementary Box 1). Thus, it is uncertain if a predicted 12% annual growth rate of the global NGS market [25] will be reflected in a similar growth of job and career opportunities in clinical molecular genetic diagnosis. In contrast, the universe of pathogenic variants at the cytogenetic level is infinite, and genotype-phenotype associations are more unpredictable because of the frequent involvement of multiple genes and the diversity of mechanisms underlying genetic disease, including clonal evolution. Consequently, job opportunities will continue to exist because in clinical cytogenomics human interventions will continue to be needed for the interpretation and reporting of pathogenic findings. It is our hope that the training programs of the future will deliver sufficient numbers of competent CLGs for accomplishing that task. Accreditation and participation in EQA schemes are crucial factors to evaluate and uphold cytogenomic competence.
References
Gilissen C, Hehir-Kwa JY, Thung DT, van de Vorst M, van Bon BW, Willemsen MH, et al. Genome sequencing identifies major causes of severe intellectual disability. Nature. 2014;511:344–7.
Caspar SM, Dubacher N, Kopps AM, Meienberg J, Henggeler C, Matyas G, et al. Clinical sequencing: from raw data to diagnosis with lifetime value. Clin Genet. 2018;93:508–19.
White SJ, Laros JFJ, Bakker E, Cambon-Thomsen A, Eden M, Leonard S. Critical points for an accurate human genome analysis. Hum Mutat. 2017;38:912–21.
Redin C, Brand H, Collins RL, Kammin T, Mitchell E, Hodge JC, et al. The genomic landscape of balanced cytogenetic abnormalities associated with human congenital anomalies. Nat Genet. 2017;49:36–45.
Hochstenbach R, van Binsbergen E, Schuring-Blom H, Buijs A, Ploos van Amstel HK. A survey of undetected, clinically relevant chromosome abnormalities when replacing postnatal karyotyping by whole genome sequencing. Eur J Med Genet. 2019;62:103543.
Nowakowska BA, de Leeuw N, Ruivenkamp CA, Sikkema-Raddatz B, Crolla JA, Thoelen R, et al. Parental insertional balanced translocations are an important cause of apparently de novo CNVs in patients with developmental anomalies. Eur J Hum Genet. 2012;20:166–70.
Silva M, de Leeuw N, Mann K, Schuring-Blom H, Morgan S, Giardino D, et al. European guidelines for constitutional cytogenomic analysis. Eur J Hum Genet. 2019;27:1–16.
Regier DS, Ferreira CR, Hart S, Hadley DW, Muenke M. Medical genetics and genomic medicine in the United States. Part 2: Reproductive genetics, newborn screening, genetic counseling, training, and registries. Mol Genet Genom Med. 2017;5:621–30.
Liehr T, Carreira IM, Aktas D, Bakker E, Rodríguez de Alba M, Coviello DA, et al. European registration process for Clinical Laboratory Geneticists in genetic healthcare. Eur J Hum Genet. 2017;25:515–9.
Zhang H, Yu J, Ming Q, Bao L, Wu B-L, Li P. On the globalization and standardization of medical genetics and genomics as clinical and laboratory specialties. North Am J Med Sci. 2014;7:194–8.
Liehr T, Carreira IM, Balogh Z, Garrido ED, Verdorfer I, Coviello DA, et al. Regarding the rights and duties of Clinical Laboratory Geneticists in genetic healthcare systems; results of a survey in over 50 countries. Eur J Hum Genet. 2019;27:1168–74.
Claustres M, Kožich V, Dequeker E, Fowler B, Hehir-Kwa JY, Miller K, et al. Recommendations for reporting results of diagnostic genetic testing (biochemical, cytogenetic and molecular genetic). Eur J Hum Genet. 2014;22:160–70.
Hastings RJ, Howell RT. The importance and value of EQA for diagnostic genetic laboratories. J Community Genet. 2010;1:11–7.
Hochstenbach R, Slunga-Tallberg A, Devlin C, Floridia G, de Alba MR, Bhola S, et al. Fading competency of cytogenetic diagnostic laboratories: the alarm bell has started to ring. Eur J Hum Genet. 2017;25:273–4.
Johansson BJ, Harrison CJ. Acute myeloid leukemia. In: Mitelman F, Helm S, editors. Cancer Cytogenetics, 4th edn. Chichester, UK: John Wiley & Sons, Ltd; 2015. pp 62–125.
Gardner RJM, Amor DJ. Gardner and Sutherland’s Chromosome Abnormalities and Genetic Counseling, 5th edn. Oxford Monographs on Medical Genetics, Oxford, UK: Oxford University Press; 2018.
Hartwig TS, Ambye L, Sørensen S, Jørgensen FS. Discordant non-invasive prenatal testing (NIPT) - a systematic review. Prenat Diagn. 2017;37:527–39.
van der Meij KRM, Sistermans EA, Macville MVE, Stevens SJC, Bax CJ, Bekker MN, et al. TRIDENT-2: national implementation of genome-wide non-invasive prenatal testing as a first-tier screening test in the Netherlands. Am J Hum Genet. 2019;105:1091–101.
ISO 15189:2012(en), Medical laboratories - requirements for quality and competence. Geneva, Switzerland: International Organization for Standardization; 2012. https://www.iso.org/obp/ui/#iso:std:iso:15189:ed-3:v2:en.
ISCN 2016. In: An international system for human cytogenomic nomenclature. McGowan-Jordan J, Simons A, Schmid M, editors. Basel, Freiburg, Switzerland: Karger; 2016.
Saunders CJ, Miller NA, Soden SE, Dinwiddie DL, Noll A, Alnadi NA, et al. Rapid whole-genome sequencing for genetic disease diagnosis in neonatal intensive care units. Sci Transl Med. 2012;4:154ra135.
Willig LK, Petrikin JE, Smith LD, Saunders CJ, Thiffault I, Miller NA, et al. Whole-genome sequencing for identification of Mendelian disorders in critically ill infants: a retrospective analysis of diagnostic and clinical findings. Lancet Respir Med. 2015;3:377–87.
Mestek-Boukhibar L, Clement E, Jones WD, Drury S, Ocaka L, Gagunashvili A, et al. Rapid Paediatric Sequencing (RaPS): comprehensive real-life workflow for rapid diagnosis of critically ill children. J Med Genet. 2018;55:721–8.
indeed.com, https://www.indeed.com/q-Clinical-Molecular-Genetics-jobs.html and https://www.indeed.com/q-Clinical-Cytogenetic-jobs.html. Accessed 5 Dec 2019.
Grandview Research. Next Generation Sequencing Market Size, Share & Trends Analysis Report By Application (HLA Typing, Consumer Genomics), By Technology, By Workflow, By End Use, By Region, And Segment Forecasts, 2020–2027. Report ID: 978-1-68038-428-4, 2020.
Tecklenburg J. Neue Musterweiterbildungsordnung Humangenetik. Medizinische Genetik. 2019;31:230–6. (in German)
Mantere T, Kersten S, Hoischen A. Long-read sequencing emerging in medical genetics. Front Genet. 2019;10:426.
Chan S, Lam E, Saghbini M, Bocklandt S, Hastie A, Cao H, et al. Structural variation detection and analysis using Bionano optical mapping. Methods Mol Biol. 2018;1833:193–203.
Neveling K, Mantere T, Vermeulen S, Oorsprong M, van Beek R, Kater-Baats E, et al. Next generation cytogenetics: comprehensive assessment of 48 leukemia genomes by genome imaging. 2020. https://doi.org/10.1101/2020.02.06.935742.
Wang H, Jia Z, Mao A, Xu B, Wang S, Wang L, et al. Analysis of balanced reciprocal translocations in patients with subfertility using single-molecule optical mapping. J Assist Reprod Genet. 2020;37:509–16.
Liehr T, Klein E, Mrasek K, Kosyakova N, Guilherme RS, Aust N, et al. Clinical impact of somatic mosaicism in cases with small supernumerary marker chromosomes. Cytogenet Genome Res. 2013;139:158–63.
Spittel H, Kubek F, Kreskowski K, Ziegler M, Klein E, Hamid AB, et al. Mitotic stability of small supernumerary marker chromosomes: a study based on 93 immortalized cell lines. Cytogenet Genome Res. 2014;142:151–60.
Dong Z, Zhang J, Hu P, Chen H, Xu J, Tian Q, et al. Low-pass whole-genome sequencing in clinical cytogenetics: a validated approach. Genet Med. 2016;18:940–8.
King DA, Sifrim A, Fitzgerald TW, Rahbari R, Hobson E, Homfray T, et al. Deciphering Developmental Disorders Study. Detection of structural mosaicism from targeted and whole-genome sequencing data. Genome Res. 2017;27:1704–14.
Biesecker LG, Spinner NB. A genomic view of mosaicism and human disease. Nat Rev Genet. 2013;14:307–20.
Hochstenbach R, Krijtenburg PJ, van der Veken LT, van der Smagt J, Roeleveld-Versteegh A, Visser G, et al. Monosomy 20 mosaicism revealed by extensive karyotyping in blood and skin cells: case report and review of the literature. Cytogenet Genome Res. 2014;144:155–62.
Hahnemann JM, Vejerslev LO. Accuracy of cytogenetic findings on chorionic villus sampling (CVS) -diagnostic consequences of CVS mosaicism and non-mosaic discrepancy in centres contributing to EUCROMIC 1986-1992. Prenat Diagn. 1997;17:801–20.
Allyse M, Minear MA, Berson E, Sridhar S, Rote M, Hung A, et al. Non-invasive prenatal testing: review of international implementation and challenges. Int J Women’s Health. 2015;16:113–26.
Minear MA, Lewis C, Pradhan S, Chandrasekharan S. Global perspectives on clinical adoption of NIPT. Prenat Diagn. 2015;35:959–67.
Phan MD, Nguyen TV, Trinh HNT, Vo BT, Nguyen TM, Nguyen NH, et al. Establishing and validating noninvasive prenatal testing procedure for fetal aneuploidies in Vietnam. J Matern Fetal Neonatal Med. 2019;32:4009–15.
Levy-Sakin M, Pastor S, Mostovoy Y, Li L, Leung AKY, McCaffrey J, et al. Genome maps across 26 human populations reveal population-specific patterns of structural variation. Nat Commun. 2019;10:1025.
Ibn-Salem J, Köhler S, Love MI, Chung H-R, Huang N, Hurles ME, et al. Deletions of chromosomal regulatory boundaries are associated with congenital disease. Genome Biol. 2014;15:423.
Spector JD, Wiita AP. ClinTAD: a tool for copy number variant interpretation in the context of topologically associated domains. J Hum Genet. 2019;64:437–43.
Middelkamp S, Vlaar JM, Giltay J, Korzelius J, Besselink N, Boymans S, et al. Prioritization of genes driving congenital phenotypes of patients with de novo genomic structural variants. Genome Med. 2019;11:79.
Acknowledgements
We wish to thank all assessors of GenQA for contributing to the results described in Table 1. We are gratefully indebted to Cheryl Guiver (GenQA) for technical assistance. We are also grateful to the colleagues who supplied information about their countries’ training program for CLGs: Kevin Carpenter (Sydney) and Angela Brown (Wellington) for Australia and New Zealand, Martina Witsch-Baumgartner (Innsbruck) for Austria, Wim Wuyts (Antwerp) for Belgium, Lotte Risom (Copenhagen) for Denmark, Nina Horelli-Kuitunen (Helsinki) for Finland, Thomas Haaf (Würzburg) for Germany, Domenico Coviello (Genova) for Italy, Olaug Rødningen (Oslo) for Norway, Maria Sąsiadek (Warsaw) for Poland, Anna Norberg (Umeå) for Sweden, Jean-Louis Blouin (Geneva) for Switzerland, Seher Başaran (Istanbul) for Turkey, and James Steer (Sheffield) for the United Kingdom. We thank Lieve Christiaens (Illumina Inc.) for inspiring discussions, and we thank the reviewers for pointing out to our possibilities to improve the clarity and conciseness of our manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Hochstenbach, R., Liehr, T. & Hastings, R.J. Chromosomes in the genomic age. Preserving cytogenomic competence of diagnostic genome laboratories. Eur J Hum Genet 29, 541–552 (2021). https://doi.org/10.1038/s41431-020-00780-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-020-00780-y
This article is cited by
-
The need for recognition of core professional groups in genetics healthcare services in Europe
European Journal of Human Genetics (2022)
-
About classical molecular genetics, cytogenetic and molecular cytogenetic data not considered by Genome Reference Consortium and thus not included in genome browsers like UCSC, Ensembl or NCBI
Molecular Cytogenetics (2021)
-
What’s new in EJHG in April
European Journal of Human Genetics (2021)
