
Abstract
The low uptake of presymptomatic testing in Huntington disease prompted us to question family members on how they handle the transmission of information regarding genetic risk. We hypothesised that in 2019, parents would inform their at-risk children about their genetic risk more and at a younger age than in 2000, given the availability of prenatal diagnosis, French legislation changes since 2011, and recent therapeutic advances. We made a questionnaire available about the transmission of genetic information within families with Huntington disease in 2000 and 2019. We obtained 443 questionnaires (295 in 2019 and 148 in 2000). Participants were mainly at-risk for Huntington disease (n = 113), affected (n = 85), and spouses (n = 154). In 2019, participants had a higher mean education level (p < 0.01) and a mean age of 44.1 ± 15.1 years (vs 48.1 ± 11.4 years in 2000, p < 0.01). They had been informed about the risk of being a carrier at around 30 years of age (29.0 ± 14.2 in 2019 vs 32.2 ± 13.8 in 2000, p = 0.09). However, they would inform at an earlier age (≤18 years, 67% vs 59%, p = 0.16). Information on transmission risk had been given primarily by parents (45% vs 30%, p = 0.06). In addition, genetic testing for relatives unaware of their status was recommended more frequently in 2019 (46% vs 32%, p < 0.001). Respondents in 2019 recommended genetic testing more often but overall attitudes towards information and testing have not changed significantly over the 19-year time period since the questionnaire was first delivered even despite recent clinical trials potential disease modifying therapies.
Similar content being viewed by others

Introduction
The debate on the justification for being tested in Huntington disease (HD [MIM: 143100]) began long before the existence of such a test. Predictive testing via linkage analysis first became available in 1986, supported by patient associations. The first recommendations were adopted in 1989 [1, 2], thus bringing to fruition a collaboration between affected families and professionals. Even after revision, the five guiding principles (“beneficence, autonomy, informed decision, confidentiality and fairness”) have remained the same [3]. Identification of the huntingtin gene (HTT [MIM: 613004]) in 1993 enabled direct testing [4]. Despite early suggestions that many at-risk individuals would ask for the test, uptake has remained low, estimated between 5 and 20% across countries [5,6,7]. Prenatal testing uptake is even lower, ~20% of carriers of reproductive age [8,9,10]. This has prompted us to investigate how HD family members pass on genetic-risk information. HD is an autosomal dominant neurodegenerative disorder, characterised by psychiatric manifestations, cognitive impairment, and abnormal motor movements, with onset of symptoms at midlife [11]. Historically, HD has been associated with stigma and taboo [12]. Individuals at risk of HD can choose whether or not to undergo genetic testing for a familial pathogenic expansion in the HTT gene following internationally recognised presymptomatic testing guidelines [2, 3]. Long-term experience acquired with this disease has served as a model in the development of presymptomatic testing protocols for other late-onset neurogenetic diseases [13, 14].
Many families believe that it is the parents’ responsibility to disclose genetic information to their children [15, 16], but parents may find themselves torn between wanting to protect their children for “as long as possible”, while knowing that they “need to be told in time to make key life decisions” [15]. Hence, the choice to delay disclosure may be perceived as a preferable option, as parents seek to protect their children while maintaining a temporary sense of control [16]. Whether there is a preferable age to disclose risk to children is debated in the literature [17, 18]. Recommendations define the legal age for testing as 18; however, minors at risk who request the test should have access to genetic counselling, support and information which should include a discussion of all of their options for dealing with being at risk [3, 19]. The legal framework for genetic testing in France has evolved since 1994 when France was one of the first countries to regulate the use of genetic testing [20]. In 2000, the law specified that the test can only be prescribed by geneticists working in a multidisciplinary team following the international guidelines [21]. The question concerning the necessity to disclose the genetic disease to the family of an affected person was first raised in 2004 [22], without application of the law. It was discussed again in 2011 [23], but legislation came into effect only in 2013 [24]. Since then, it has been mandatory to inform relatives of a genetic disease in the family when there are preventive or curative options [23,24,25]. It is a valid concern that the absence of information regarding their carrier risk would deprive relatives of the opportunity to access genetic counselling or prenatal diagnosis. Attitudes are expected to change with the emergence of therapeutic trials. Promising methods such as lowering the level of mutant huntingtin, either by targeting mutant HTT DNA or RNA [26,27,28], and clinical trials with antisense therapy are currently underway (NCT03761849, NCT03225846, NCT03225833, NCT04120493).
In 2000, we asked about the informing of genetic risk within families with HD and found that only 30% of the respondents had been informed about their risk by their parents and 24% had found out by themselves. This result was puzzling, because we would have expected a much higher rate of communication concerning this familial disease. In light of new therapeutic avenues, access to prenatal diagnosis, and recent legislation, we aimed to investigate possible changes of attitudes towards disclosure of familial information and predictive testing after 19 years.
Methods
Participants and questionnaires
Questionnaires were distributed in 2000 and 2019. They were composed of 18 questions (Tables 1 and 2): demographic questions concerning the sex of the respondents, their age, their number of children and their educational level; questions regarding their experience with genetic information and testing: their status, their age when they became aware of HD, the person who informed them (for the spouses the responses concerned their partner); questions regarding their attitudes towards information and testing: what would be the ideal age to inform children of their risk and who should do this, should presymptomatic testing be offered regardless of age, even under 18 years of age, and the reasons why they answered yes or no. The questions regarding the motivations were conditional branching open-ended questions, their answers were prioritised according to importance and only the first priority was considered. In 2019, one question concerning the 2011 law was added. The complete questionnaire is provided in the supplementary data.
In 2000, the questionnaire was first distributed via a French HD family support organisation (Association Huntington France) through their newsletter. Respondents were invited to send the anonymous questionnaire back via postal mail. In 2019, individuals were invited to answer an online anonymous questionnaire via REDCap (Research Electronic Data Capture Vanderbilt University, Nashville, Tennessee, USA) [29, 30]. We collected and managed the study data and the database was hosted at the ICM (Paris Brain Institute). The questionnaire was accessible via two HD family support websites (Association Huntington France and Huntington Espoir), and was also made available after genetic counselling sessions in the Genetics Department at the Pitié-Salpêtrière University Hospital. The following groups of participants were established by self-declaration in the first questions of the questionnaire: patients with symptoms of HD and genetic confirmation, at-risk individuals (children, siblings of an affected patient, not knowing their genetic status), asymptomatic carriers of the pathological repeat expansion (without signs of HD yet but knowing their genetic status after a presymptomatic test), non-carriers of the pathological repeat expansion (knowing their genetic status after a presymptomatic test, thus certain not to develop HD) and spouses of all the above-mentioned groups (Table 1). All were legal adults.
According to French legislation, as this study relies only on surveys and interviews, it does not require specific approval by an ethics committee [31, 32]. Written consent forms were signed by all participants, and we declared the study to be compliant with the reference methodology to the National Commission for Data protection and Individual rights (n° 2131944v0, December 15th, 2017).
Statistical analysis
Data are reported as frequencies (per cent) for qualitative variables and the mean ± SD for quantitative variables. Chi-square tests or Student t tests were used to compare the 2000 and 2019 answers, depending on the nature of the variables. Results were considered to be statistically significant for p ≤ 0.05. Statistical analysis was performed using SAS software (V.9.4; SAS Institute, Cary, North Carolina, USA).
Results
We collected 295 and 148 complete questionnaires in 2019 and 2000, respectively, and analysed a total of 443 questionnaires, the status of the respondent was unknown for two questionnaires. The spouses group was composed as follows: spouses of HD patients (n = 65 in 2019, n = 50 in 2000), spouses of at-risk individuals (n = 14 in 2019, n = 8 in 2000), spouses of presymptomatic pathogenic expansion carriers (n = 14 in 2019, n = 1 in 2000), spouses of non-carriers of the pathogenic expansion (n = 1 in 2019, n = 1 in 2000). The groups were distributed differently between the two timepoints (p < 0.01): the largest group was spouses (32% vs 41%), followed by at-risk individuals (22% vs 32%), HD patients with genetic confirmation (21% vs 16%), presymptomatic pathogenic expansion carriers (16% vs 6%), and non-carriers of the pathogenic expansion (9% vs 5%) for 2019 and 2000, respectively (Table 1).
In 2019, participants were younger (44.1 ± 15.1 years vs 48.1 ± 11.4 years, p < 0.01). There were relatively more women than men participating at both timepoints (67% in 2019 vs 66% in 2000, p = 0.83). In 2019, participants had a higher level of education (p < 0.01) and fewer children (p < 0.001).
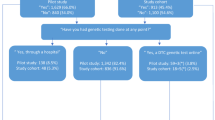
In 2019, parents were favourably designated as the major informants (58% vs 47% in 2000, p = 0.01), with less involvement of medical teams (35% vs 50%). Nonetheless, only 45% of participants had been informed by their parents, which is more than in 2000 but not statistically different (30%, p = 0.06) (Table 2, Fig. 1). A large portion of the participants believed that offspring should be informed before or when they reach the legal age of adulthood (18 years) (67% in 2019 vs 59% in 2000, p = 0.16).

A. Questions and responses about the informant. B. Opinions about who should be the informant. C. Attitudes towards presymptomatic testing regardless of age. D. Attitudes towards presymptomatic testing regarding minors.
Opinions about presymptomatic testing have changed over time: in 2019, it was more highly encouraged (46% vs 32% in 2000, p < 0.001) even before 18 years of age (28% vs 14% in 2000, p < 0.01). In parallel, fewer respondents were opposed to the test, regardless of age, even before the age of 18 (27% vs 57.5% in 2000, p < 0.001; and 47% vs 63% in 2000, p < 0.01, respectively). In 2019, more participants questioned whether to recommend testing or not (27% vs 10.5% in 2000, p < 0.001).
The motivations to recommend testing were not significantly different between 2019 and 2000 (p = 0.097): the “right to know” about their status and non-transmission to offspring were important concerns at both timepoints (52% in 2019 vs 26% in 2000 for the “right to know”, and 24% in 2019 vs 35% in 2000 for “to prevent transmission”). Another reported motivation for undertaking testing was to anticipate and plan for the future (19% in 2019 and 29% in 2000). To benefit from treatment was the least-cited reason in 2019, as in 2000 (5% and 10%, respectively). The reasons given by respondents who did not advise testing were not significantly different in 2019 and 2000 (p = 0.69). The most prevalent reason given was a concern for individual choice (48% vs 53%). The second was the potential for negative psychological impact (34% vs 35%). The absence of a cure also discouraged some (18% vs 12%).
The motivations to recommend testing before age 18 were significantly different (p < 0.001) from 2000 to 2019. The “right to know” was the main argument for testing minors in 2019 (45%), contrary to in 2000 (25%). Concerns about the future justified testing in children for nearly half of the respondents who were in favour (40.5% in 2019 vs 17% in 2000). Eliminating parental uncertainty about their child’s status, the first reason given in 2000 (50%), was a modest concern in 2019 (6.5%). The hope for a cure was a relatively minor motivation (8% for both years). The main reasons against the testing of minors (p = 0.16) were the potential for negative consequences (71% and 65%), the concern for children’s autonomy in choosing to test or not (19% and 15%), and the absence of available treatment (10% and 20%) in 2019 and 2000, respectively.
Only 13% of the respondents had heard about the change in the French legal framework for the disclosure of genetic information to kin.
Discussion
Our study is the first to compare how information about genetic risk for HD and testing is disclosed within families two decades apart.
We hypothesised that, in 2019, respondents would be more prone to undergo or recommend testing, especially since the prevalence of HD is higher than previously expected [33, 34]. This was found to be true, as more respondents recommended testing in 2019 than in 2000, even if the absolute numbers did not exceed half of the respondents. This low value could explain the stability of testing uptake, between 5 and 25%, depending on the country [5, 6, 25]. Similarly, we have reported consistently low uptake of predictive testing for HD in France, as well as for inherited cerebellar ataxias and Creutzfeldt-Jakob disease [35, 36]. The number of tests, when reported, has a tendency to decrease [5, 37] with rates of withdrawal from testing that increase over time [38]. The overall population seeking testing has remained comparable, with a mean age of 35–37 years and a tendency for more requests from women [5, 38, 39].
We also hypothesised that in 2019, parents would inform their children about their genetic risk more frequently. Despite the fact that parents were designated as the preferred informants both in 2019 and 2000, less than half of participants had been informed by their parents in 2019, higher but not statistically different from 2000. However, there is a trend towards a larger proportion being informed by their parents.
Our hypothesis about information being transmitted earlier in more recent years has also been invalidated. There have been no extensive studies to determine the age at which individuals find out that they are at risk for HD. We previously reported that 25% at-risk individuals had been informed earlier than 50% at-risk ones, at a mean age of 24 years, but this result must be put into perspective, given the small sample size [40]. This may imply that parents do not disclose genetic risk unless they have sufficient motivation to do so. The right time to tell children may be prior to their own family planning [15] or linked to important life stages [18]. Around 60% of study participants self-reported that children should be informed about their risk before or at the age of 18 years, in 2000 and in 2019. This is intriguing, as the mean age at which they themselves were informed about the genetic risk in their own families was in their thirties in 2000 and it was still the case in 2019. One could imagine that for affected or presymptomatic parents, informing children about their risk induces a feeling of guilt, leading them to delay or avoid sharing information. The fear of hostility from children towards the transmitting parent could partially explain this result. Spouses, however, are not exempt from this culpability, especially as they sometimes decide to have children with knowledge of the risk. Unfortunately, we could not examine differences in attitudes between spouses and other groups given the small sample sizes.
In this study, we aimed to evaluate the evolution in motivations to test. In 2000, the main motivation to have the test was to prevent transmission and thus it was driven by a concrete action: refrain from having children or explore the options for prenatal diagnosis and preimplantation genetic diagnosis. In 2019, this was no longer the main motivation. Respondents highlighted the right to know, in accordance with the literature, in which the most common patient-reported reason for undergoing predictive testing is to reduce uncertainty [5, 39]. Testing would thus enable them to feel more autonomous. Although respondents do not cite the potential for treatment as a strong motivation for testing, huge progress in understanding the mechanisms of HD have perhaps given rise to hope. Parents may now think that during the time before their child reaches adulthood, a cure will have been discovered, therefore contributing to a shift in attitudes. Current reasons for testing before the age of 18 highlight a desire for children’s autonomy (“they have right to know”, “to anticipate and build the future”), in contrast with results from 2000, when the focus was more on the parents (“to remove doubt from the parents”). Guidelines and position papers recommend that testing should be delayed until adulthood [19, 41], when young adults have reached their full intellectual and psychosocial capacities. This would allow them to make an autonomous choice, as opposed to one driven by their parents’ wishes or fears. There has been a clear drop in the proportion of respondents resolutely opposed to testing since 2000. Individuals are less prone to decline testing and more open to assuaging their doubt. Uncertainty about the appropriateness of the test before or after 18 years of age is still an indisputable reality.
Given the French legislation and increased access to information in 2019, one might have expected a much higher rate of informing children of the risk of transmission. Parents may try to protect their children from a harmful reality and perhaps protect themselves from the feeling of guilt of transmission. Indeed, most respondents consistently argued that testing is an individual choice that could potentially have negative psychological consequences. This autonomous decision depends on maturity, life experience and temperament, which may or may not provide the ability to cope with the result. Thus, “the autonomy of each family member regarding his/her decision to request further information should be respected” [25]. The awareness of carrier status can be a burden, for which the benefit is not always clear. If the result is unfavourable, uncertainty about the age at onset begins. This situation results in pre-manifest carriers actively observing themselves and possibly misinterpreting events as indicators of disease onset, effectively “feeling sick before being sick” [14]. In addition, a favourable result may not produce the expected relief. This emotional experience could be comparable to survivor’s guilt [13, 42, 43]. Non-carriers may feel guilty towards their carrier siblings and this information can disrupt family relationships and accentuate tensions [44]. Moreover, the test result can have a profound impact on one’s identity. It can be a long process to recover from being at risk and reorganise one’s life without the threat of disease [25]. Interestingly, the fear of a negative psychological impact is even stronger for testing before 18 years of age: children are considered to be less able to cope with an unfavourable result or, perhaps more likely, their parents wish to protect them (perhaps overly so) (“he is too young to worry about that”).
We might assume that prenatal options, for which there is now broad access, would be a strong and sufficient reason for testing. However, it is rarely requested, as only ~20% of carriers of reproductive age request prenatal or preimplantation diagnosis [8,9,10, 45]. This could be explained by a reluctance to terminate pregnancy, which may be intolerable in the context of a late-onset disease [46]. Moreover, it could call into question the value of one’s own life, and more broadly whether life with HD is worth living. Surprisingly, children born after a prenatal diagnosis are only informed late in their childhood about their favourable genetic status, at around 12–14 years of age [8].
With the advent of potentially disease modifying therapies [26,27,28], it is legitimate to question whether this would motivate parents to inform their children more or at a younger age. However, only a minority considered potential treatment as a motivation for testing, even in 2019. This result is surprising but consistent with those found in the literature [5, 39]. We can infer that participants may not be hopeful, or perhaps have not been informed about new therapeutic strategies currently under development. The burden of a familial illness affecting many of its members and the perception of the seriousness of neurological disorders probably take precedence over hope for effective treatments. These findings reaffirm our current practice of providing systematic explanations of therapeutic approaches and clinical trials during consultations in our centre and encourage us to enhance these explanations.
Finally, a vast majority were not familiar with the content of the change in French legal framework for the disclosure of genetic information to kin. The clinician must inform the person requesting the test of the law and explain the risks that would arise from the patient’s failure to disclose. If the patient does not want to disclose the risk to his/her family him/herself, the clinician can send an anonymous letter inviting relatives to contact a genetics department [47]. In case of opposition, the decision of the patient must be recorded in the medical file [25]. Support groups of patients affected by the disease consider the legislature to be insensitive to the suffering of people who have just learned they are carriers by adding the considerable burden of being obliged to pass on this information to their relatives [48]. Thus, the law is not well adapted to late-onset diseases for which the only prevention is prenatal or preimplantation diagnosis. A few questions remain open: should a patient be held legally responsible if he/she fails to disclose information about disease to his/her relatives, given that there are neither preventive measures nor curative treatments, and can we assume that prenatal or preimplantation diagnosis are compelling reasons? These options concern only potential future births, and it can be argued that the negative impact of disclosure exceeds the potential for positive outcomes calling into question the benefit of such information. Furthermore, the obligation to inform conflicts with one of the five guiding principles for presymptomatic testing: confidentiality [3]. In the case of this familial disease, the autonomy of each family member to decide whether to request further information should be respected.
Our study has several limitations. Firstly, the small number of respondents per group did not allow for any subgroup analysis, we are thus unable to compare responses according to group status. Secondly, the reliance on self-declared disease status could introduce group misattribution due to anosognosia and/or denial. Finally, there may be selection bias as participants were self-referred, skewing the sample towards more involved participants as they are engaged in the genetic counselling process.
A strength of our study is its utility in guiding our reflection on improving genetic counselling and support for the families concerned. After our survey in 2000, our team began work on a book to help children and parents face the genetic disease and to promote familial discussion [49]. Furthermore, based on our study findings risk disclosure to relatives is thoroughly discussed during pre-test sessions with a psychologist, and after the result has been received. We also receive children in consultation to help explain the familial disease, if needed. We plan to reinforce our work before and after predictive testing within our interdisciplinary team, to help families to manage what’s often perceived as a “time bomb”. Feedback about our study from HD patient organizations will help us to explore new potential projects together. We have already developed support groups for helpers and for presymptomatic carriers, and now aim to create a group focused on strategies to inform children and relatives.
In conclusion, overall attitudes towards testing in 2019 have not changed radically since 2000 and are probably unlikely to until disease modifying therapy becomes a reality. It would be of interest to conduct a new survey when such a treatment becomes available.
References
World Federation of Neurology: Research Committee. Research Group on Huntington’s chorea. Ethical issues policy statement on Huntington’s disease molecular genetics predictive test. J Neurol Sci. 1989; 94: 327–32.
International Huntington Association (IHA) and the World Federation of Neurology (WFN) Research Group on Huntington’s Chorea. Guidelines for the molecular genetics predictive test in Huntington’s disease. Neurology. 1994;44:1533–1533.
MacLeod R, Tibben A, Frontali M, Evers-Kiebooms G, Jones A, Martinez-Descales A, et al. Recommendations for the predictive genetic test in Huntington’s disease: Recommendations for the predictive genetic test in Huntington’s disease. Clin Genet. 2013;83:221–31.
A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. The Huntington’s Disease Collaborative Research Group. Cell 1993; 72: 971–83.
UK Huntington’s Disease Prediction Consortium, Baig SS, Strong M, Rosser E, Taverner NV, Glew R, et al. 22 Years of predictive testing for Huntington’s disease: the experience of the UK Huntington’s Prediction Consortium. Eur J Hum Genet. 2016;24:1396–402.
Panas M, Karadima G, Vassos E, Kalfakis N, Kladi A, Christodoulou K, et al. Huntington’s disease in Greece: the experience of 14 years. Clin Genet. 2011;80:586–90.
Morrison P, Harding-Lester S, Bradley A. Uptake of Huntington disease predictive testing in a complete population. Clin Genet. 2011;80:281–6.
Bouchghoul H, Clément S-F, Vauthier D, Cazeneuve C, Noel S, Dommergues M, et al. Prenatal testing in Huntington disease: after the test, choices recommence. Eur J Hum Genet. 2016;24:1535–40.
on behalf of the UK HD Predictive Testing Consortium, Piña-Aguilar RE, Simpson SA, Alshatti A, Clarke A, Craufurd D, et al. 27 years of prenatal diagnosis for Huntington disease in the United Kingdom. Genet Med. 2019;21:1639–43.
van Rij M, de Koning Gans P, Aalfs C, Elting M, Ippel P, Maat-Kievit J, et al. Prenatal testing for Huntington’s disease in the Netherlands from 1998 to 2008: Prenatal testing for Huntington’s disease. Clin Genet. 2014;85:78–86.
Bates GP, Dorsey R, Gusella JF, Hayden MR, Kay C, Leavitt BR, et al. Huntington disease. Nat Rev Dis Prim. 2015;1:15005.
Vamos M, Hambridge J, Edwards M, Conaghan J. The Impact of Huntington’s Disease on Family Life. Psychosomatics. 2007;48:400–4.
Gargiulo M, Lejeune S, Tanguy M-L, Lahlou-Laforêt K, Faudet A, Cohen D, et al. Long-term outcome of presymptomatic testing in Huntington disease. Eur J Hum Genet. 2009;17:165–71.
Gargiulo M, Tezenas du Montcel S, Jutras MF, Herson A, Cazeneuve C, Durr A. A liminal stage after predictive testing for Huntington disease. J Med Genet. 2017;54:1–520.
Forrest K, Simpson S, Wilson B, Van Teijlingen E, McKee L, Haites N, et al. To tell or not to tell: barriers and facilitators in family communication about genetic risk: Family communication about genetic risk. Clin Genet. 2003;64:317–26.
Wilson BJ, Forrest K, van Teijlingen ER, McKee L, Haites N, Matthews E, et al. Family Communication about Genetic Risk: the Little That Is Known. Public Health Genom. 2004;7:15–24.
Holt K. What Do We Tell the Children? Contrasting the Disclosure Choices of Two HD Families Regarding Risk Statusand Predictive Genetic Testing. J Genet Couns. 2006;15:253–65.
Klitzman R, Thorne D, Williamson J, Chung W, Marder K. Disclosures of Huntington disease risk within families: patterns of decision-making and implications. Am J Med Genet. 2007;143A:1835–49.
Joly L, Thauvin-Robinet C, Huet F, Pinoit J-M, Contrain A, Cassini C, et al. Les tests génétiques présymptomatiques chez le mineur: enquête auprès des généticiens français et position du groupe français de génétique prédictive. Arch de Pédiatrie. 2010;17:1000–7.
Loi n° 94-654 du 29 juillet 1994 relative au don et à l’utilisation des éléments et produits du corps humain, à l’assistance médicale à la procréation et au diagnostic prénatal. 1994 https://www.legifrance.gouv.fr/jorf/id/JORFTEXT000000549618/ (accessed 22 Sep 2020).
Décret n° 2000-570 du 23 juin 2000 fixant les conditions de prescription et de réalisation des examens des caractéristiques génétiques d’une personne et de son identification par empreintes génétiques à des fins médicales et modifiant le code de la santé publique (deuxième partie: décrets en Conseil d’Etat). 2000 https://www.legifrance.gouv.fr/jorf/id/JORFTEXT000000216521/ (accessed 19 Sep 2020).
Loi n° 2004-800 du 6 août 2004 relative à la bioéthique. 2004 https://www.legifrance.gouv.fr/loda/id/JORFTEXT000000441469/2020-09-21/ (accessed 22 Sep 2020).
Loi n° 2011-814 du 7 juillet 2011 relative à la bioéthique (1). 2011. https://www.legifrance.gouv.fr/jorf/id/JORFTEXT000024323102/ (accessed 22 Sep 2020).
Décret n° 2013-527 du 20 juin 2013 relatif aux conditions de mise en œuvre de l’information de la parentèle dans le cadre d’un examen des caractéristiques génétiques à des fins médicales. 2013. https://www.legifrance.gouv.fr/jorf/id/JORFTEXT000027592003/ (accessed 22 Sep 2020).
Clément S, Gargiulo M, Feingold J, Durr A. Guidelines for presymptomatic testing for Huntington’s disease: past, present and future in France. Rev Neurologique. 2015;171:572–80.
Wild EJ, Tabrizi SJ. Therapies targeting DNA and RNA in Huntington’s disease. Lancet Neurol. 2017;16:837–47.
Tabrizi SJ, Ghosh R, Leavitt BR. Huntingtin Lowering Strategies for Disease Modification in Huntington’s Disease. Neuron. 2019;101:801–19.
Tabrizi SJ, Leavitt BR, Landwehrmeyer GB, Wild EJ, Saft C, Barker RA, et al. Targeting Huntingtin Expression in Patients with Huntington’s Disease. N Engl J Med. 2019;380:2307–16.
Harris PA, Taylor R, Thielke R, Payne J, Gonzalez N, Conde JG. Research electronic data capture (REDCap)—A metadata-driven methodology and workflow process for providing translational research informatics support. J Biomed Inform. 2009;42:377–81.
Harris PA, Taylor R, Minor BL, Elliott V, Fernandez M, O’Neal L, et al. The REDCap consortium: building an international community of software platform partners. J Biomed Inform. 2019;95:103208.
Loi n° 2012-300 du 5 mars 2012 relative aux recherches impliquant la personne humaine (1). 2012 https://www.legifrance.gouv.fr/jorf/id/JORFTEXT000025441587 (accessed 21 Sep 2020).
Décret n° 2017-884 du 9 mai 2017 modifiant certaines dispositions réglementaires relatives aux recherches impliquant la personne humaine. 2017 https://www.legifrance.gouv.fr/jorf/id/JORFTEXT000034634217/ (accessed 21 Sep 2020).
Fisher ER, Hayden MR. Multisource ascertainment of Huntington disease in Canada: Prevalence and population at risk: Multisource Ascertainment of BC HD Patients. Mov Disord. 2014;29:105–14.
for GEMO, Thion MS, Tézenas du Montcel S, Golmard J-L, Vacher S, Barjhoux L, et al. CAG repeat size in Huntingtin alleles is associated with cancer prognosis. Eur J Hum Genet. 2016;24:1310–5.
Goizet C, Lesca G, Durr A. Presymptomatic testing in Huntington’s disease and autosomal dominant cerebellar ataxias. Neurology. 2002;59:1330–6.
Schwartz M, Brandel J-P, Babonneau ML, Boucher C, Schaerer E, Haik S, et al. Genetic Testing in Prion Disease: psychological Consequences of the Decisions to Know or Not to Know. Front Genet. 2019;10:895.
Bernhardt C, Schwan A-M, Kraus P, Epplen JT, Kunstmann E. Decreasing uptake of predictive testing for Huntington’s disease in a German centre: 12 years’ experience (1993–2004). Eur J Hum Genet. 2009;17:295–300.
Ramond F, Quadrio I, Le Vavasseur L, Chaumet H, Boyer F, Bost M et al. Predictive testing for Huntington disease over 24 years: Evolution of the profile of the participants and analysis of symptoms. Mol Genet Genomic Med. 2019; 7. https://doi.org/10.1002/mgg3.881.
Holman MA, Quillin J, York TP, Testa CM, Rosen AR, Norris VW. The Changing Age of Individuals Seeking Presymptomatic Genetic Testing for Huntington Disease. J Genet Couns. 2018;27:1157–66.
Bonnard A, Herson A, Gargiulo M, Durr A. Reverse pre-symptomatic testing for Huntington disease: double disclosure when 25% at-risk children reveal the genetic status to their parent. Eur J Hum Genet. 2019;27:22–7.
on behalf of the Public and Professional Policy Committee (PPPC) of the European Society of Human Genetics (ESHG), Borry P, Evers-Kiebooms G, Cornel MC, Clarke A, Dierickx K. Genetic testing in asymptomatic minors: background considerations towards ESHG Recommendations. Eur J Hum Genet. 2009;17:711–9.
Tibben A, MV-VD Vlis, Skraastad MI, et al. MF et al. DNA-Testing for Huntington’s disease in The Netherlands: a retrospective study on psychosocial effects. Am J Med Genet. 1992;44:94–9.
Evers-Kiebooms G, Decruyenaere M. Predictive testing for Huntington’s disease: a challenge for persons at risk and for professionals. Patient Educ Counseling. 1998;35:15–26.
Rivera-Navarro J, Cubo E, Mariscal N. Analysis of the Reasons for Non-Uptake of Predictive Testing for Huntington’s Disease in Spain: a Qualitative Study. J Genet Couns. 2015;24:1011–21.
Decruyenaere M, Evers-Kiebooms G, Boogaerts A, Philippe K, Demyttenaere K, Dom R, et al. The complexity of reproductive decision-making in asymptomatic carriers of the Huntington mutation. Eur J Hum Genet. 2007;15:453–62.
Evers-Kiebooms G, Nys K, Harper P, Zoeteweij M, Dürr A, Jacopini G, et al. Predictive DNA-testing for Huntington’s disease and reproductive decision making: a European collaborative study. Eur J Hum Genet. 2002;10:167–76.
Arrêté du 20 juin 2013 fixant le modèle de lettre adressée par le médecin aux membres de la famille potentiellement concernés en application de l’article R. 1131-20-2 du Code de la santé publique. 2013 https://www.legifrance.gouv.fr/loda/id/JORFTEXT000027592025/2020-09-21/ (accessed 21 Sep 2020).
Maladie de Huntington et information de la parentèle | editorial | Espace éthique/Ile-de-France. https://www.espace-ethique.org/ressources/editorial/maladie-de-huntington-et-information-de-la-parentele (accessed 7 Jun 2020).
Barbara D, Mansot F. Le voyage de Luna. Actes Sud/Junior: Paris; 2002.
Acknowledgements
We are very grateful to all participants who answered the questionnaires. We would like to very sincerely thank the Association Huntington France, Huntington Espoir and ICS Dingdingdong for their continuous support.
Funding
French Agency of Biomedicine (Grant n° AOR 2017-36).
Author information
Authors and Affiliations
Contributions
LP: analysis and interpretation of data and drafting/revising the paper. JH: analysis and interpretation of data and revising the paper. STdM: study concept and design, analysis and interpretation of data and revising the paper. GC, AH: acquisition of data and revising the paper. ES, AH, EP: acquisition of data. MG, AD: study concept and design, acquisition, analysis and interpretation of data, obtaining funding, study supervision and coordination and drafting/revising the paper.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Pierron, L., Hennessy, J., Tezenas du Montcel, S. et al. Informing about genetic risk in families with Huntington disease: comparison of attitudes across two decades. Eur J Hum Genet 29, 672–679 (2021). https://doi.org/10.1038/s41431-020-00776-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-020-00776-8
This article is cited by
-
Genetic counselling and testing for neurodegenerative disorders using a proposed standard of practice for ALS/MND: diagnostic testing comes first
European Journal of Human Genetics (2022)
