
Abstract
Haemophilia A and B are X-linked hemorrhagic disorders caused by gene variants in the F8 and F9 genes. Due to recessive inheritance, males are affected, while female carriers are usually asymptomatic with a wide range of factor VIII (FVIII) or IX (FIX) levels. Bleeding tendency in female carriers is extremely variable and may be associated with low clotting factor levels. This could be explained by F8 or F9 genetic variations, numerical or structural X chromosomal anomalies, or epigenetic variations such as irregular X chromosome inactivation (XCI). The aim of the study was to determine whether low FVIII or FIX coagulant activity in haemophilia carriers could be related to XCI and bleeding symptoms. HUMARA assay was performed on 73 symptomatic carriers with low clotting activity ≤50 IU/dL. Bleeding Assessment Tool (BAT) from the International Society on Thrombosis and Haemostasis (ISTH) was used to describe symptoms in the cohort of carriers. In 97% of haemophilia carriers, a specific gene variant in heterozygous state was found, which alone could not justify their low FVIII or FIX levels (≤50 IU/dL). A statistical association between XCI pattern and FVIII and FIX levels was observed. Moreover, female carriers with low coagulant activity (≤20 IU/dL) and high degree of XCI ( ≥ 80:20) had a higher ISTH-BAT score than the carriers with the opposite conditions (>20 IU/dL and <80:20). In our cohort of haemophilia carriers, XCI was significantly skewed, which may contribute to the low expression of clotting factor levels and bleeding symptoms.
Similar content being viewed by others

Introduction
Haemophilia A (OMIM:306700) and B (OMIM:306900) are the most recognised and common hereditary hemorrhagic disorders caused by gene variants in factor VIII (F8) or factor IX (F9) genes, located on the long arm of the X chromosome. Factor VIII (FVIII) and factor IX (FIX) are two plasma glycoproteins that play an essential role in coagulation. Their deficiency or dysfunction causes a defect in clot formation and consequent bleeding diathesis. Recurrent bleeding events occur spontaneously within muscles and joints, mainly in patients with severe haemophilia, resulting in disabling musculoskeletal damage and chronic arthropathy [1, 2].
Both types of haemophilia share the same manner of hereditary transmission, as a recessive X-linked trait. Thus, men are generally affected, while female carriers are usually asymptomatic and express about half the normal activity level of the factors, due to a variety of genetic defects in heterozygous state [3, 4]
Carriers of haemophilia are known to have a wide range of clotting factor levels, from very low, resembling affected males, to values within the normal range, with variable bleeding symptoms. The phenotypic expression of the disease in females may result from different genetic mechanisms, due to the presence of F8 or F9 defects in homozygous (e.g., consanguineous) or compound heterozygous state, from X chromosome abnormalities such as monosomy X (45 X, Turner syndrome), or from skewed X chromosome inactivation in heterozygous female carriers [5].
X chromosome inactivation (XCI) is an epigenetic phenomenon that occurs early in embryonic life at the blastocyst stage, in which both copies of the X chromosome (in female mammals) have an equal chance of becoming inactive. XCI is widely believed to be a random and unbiased process, but recent data reveal that quantitatively biased inactivation patterns occur in some placental mammal species. For certain X-linked disorders in humans, a secondary cell selection occurs after an initially random XCI [6, 7].
The process of inactivation of the X chromosome begins with its packaging in a transcriptionally inactive structure called heterochromatin and CpG hypermethylation, which results in the silencing of the majority of genes on one of the two X chromosomes [8, 9]. This process ensures that female XX cells have an equal dosage of X chromosomal gene products compared to male XY cells. The XCI ratio of normal females can range from a 50:50, in which each X chromosome is active in an equal number of cells, to a highly skewed ratio of 0:100, in which the same X chromosome is active for all cells [10].
In carriers of X‐linked disorders, a non-random X chromosome inactivation could result in a symptomatic female, in whom the normal X chromosome is predominantly inactive [11, 12].
In this study, we analyzed the distribution of the X inactivation pattern in a group of haemophilia carriers with coagulant factor levels of ≤50 IU/dL. In addition, bleeding tendency was quantitatively evaluated by Bleeding Assessment Tool (BAT) from the International Society on Thrombosis and Haemostasis (ISTH) and correlated with coagulation factor level and the X chromosome inactivation pattern observed in our cohort of haemophilia carriers.
Materials and methods
Subjects
We genetically characterized a total of 468 patients with mild/moderate or severe haemophilia A or haemophilia B, from 419 unrelated families, who referred to Angelo Bianchi Bonomi Haemophilia and Thrombosis Centre in Milan, Italy. Of the 244 haemophilia carriers identified at our centre from 2001 to 2019, 215 are carriers of haemophilia A and 29 of haemophilia B, with a coagulant factor level between 1 and 110 IU/dL. Among these women, we selected female carriers with a level of FVIII and FIX between 1 and 50 IU/dL, who made up 30% (73/246) of the female carriers referred to our centre [13, 14]. World Federation of Hemophilia (WFH) guidelines classify female carriers with clotting factor levels of ≤50 IU/dL as having haemophilia [15].
Seventy-nine female non-carriers of haemophilia were recruited as a control group.
The median age of carriers and non-carriers was 44 years (IQR:35–52) and 40 years (IQR:31–48), respectively.
Written informed consent was obtained from all subjects at the time of blood sample collection and was carried out in accordance with the Declaration of Helsinki.
Coagulation assays
Blood samples were collected in 3.2% buffered citrate solution (1:9 anticoagulant to whole blood). FVIII and FIX coagulant activity (FVIII:C and FIX:C) were measured using one-stage clotting assay as described in the literature [16].
FVIII and FIX activity was entered in medical records and in case of several different levels, the baseline level, defined as the lowest ever recorded value considered accurate, was provided [17].
Von Willebrand factor antigen (VWF:Ag), von Willebrand factor ristocetin cofactor activity (VWF:RCo) and FVIII binding to VWF were measured to rule out von Willebrand disease type 2 Normandy in symptomatic female carriers with low FVIII:C levels. These assays were carried out using previously described methods [18,19,20].
DNA extraction and FVIII and FIX gene analysis
Genomic DNA was extracted from whole blood according to standard procedure [21]. Analysis for the presence of intron 22 [22] and intron 1 [23] inversions of the F8 gene were performed as described in the literature. Coding regions, intron/exon boundaries, and the 5’ and 3’ untranslated regions of the F8 (NG_011403.1) and F9 (NG_007994.1) were amplified by PCR and sequenced on an ABI PRISM 3130™ Genetic Analyzer capillary sequencer (Applied Biosystems).
The F8 and F9 gene sequences were aligned using the Basic Local Alignment Search Tool (http://blast.ncbi.nlm.nih.gov). All oligonucleotides and PCR conditions are available on request.
Multiplex ligation-dependent probe amplification (MLPA) was carried out with SALSA probemix P178 and P207 (MRC Holland), according to the manufacturer’s recommendations. Amplification products were analyzed on an AB1 PRISM 3130™ Genetic Analyzer (Applied Biosystems) and the dosage quotient was calculated with COFFALYSER software (MRC Holland).
The F8 and F9 gene variants are reported according to the guidelines of the Human Genome Variation Society (http://www.hgvs.org/mutnomen/recs.html). Genetic variants were checked using the Mutalyzer website (https://mutalyzer.nl/). GeneBank accession numbers NM_000132.3 and NP_000123.1 for F8 and NM_000133.3 and NP_000124 for F9 were used as references.
ABO genotyping
ABO blood group information was collected by clinicians or through ABO blood group polymorphism analysis. Briefly, genomic DNA was amplified using primers specifically designed for exon 6 of ABO glicosyl transferase locus (NC_000009.12); 5 µl of PCR products were digested with KpnI restriction enzyme [24] and analyzed as previously described [25].
X chromosome inactivation assay
X inactivation status was studied by methylation-sensitive endonuclease digestion (HhaI), coupled with PCR analysis of a polymorphic CAG repeat in exon 1 of the human androgen receptor (HUMARA) gene (NC_000023.11) [26]. The test was performed in duplicate in the 70% and in triplicate in 30% of the analyzed samples.
The degree of the XCI pattern was calculated as peak area of [(XC1digested/non digested)/(XC2 digested/non digested) + (XC1 digested/non digested)] × 100 and was classified as random (ratios 50:50 < 80:20), skewed (ratios 80:20 < 90:10), or extremely skewed (≥90:10) [9].
In addition, one digested and one undigested male sample were included as controls in every batch of samples, to verify the completed digestion and amplification efficacy.
To discriminate whether the mutant allele (F8 or F9) is predominantly active, the affected male was analyzed, when available.
Bleeding symptoms
Bleeding tendency in female carriers of haemophilia was evaluated using the standardized, quantitative ISTH-BAT score [27]. The questionnaire can translate the severity of a range of bleeding symptoms into a final, summative bleeding score by assigning one point for every type of bleeding episode plus up to four additional points per type, depending on its nature (spontaneous versus post-traumatic or following surgical procedures), severity, duration, localization, and frequency, which were predefined for each bleeding episode [27].
Sixty-five carriers of haemophilia A and B were interviewed by a single physician, retrospectively. Bleeding data were not available for eight female carriers. A normal range for bleeding severity score was published in 2014 and the cut-off for a positive or abnormal bleeding score was set at ≥6 in adult females [28].
Statistical analysis
Data are presented as medians and interquartile ranges (IQRs) for continuous variables and as counts and percentages for categorical variables. Correlation between the percentage of X inactivation pattern and coagulant factor level was demonstrated using the Pearson method.
Fisher’s exact test was used to determine if there was a non-random association between two categorical variables (XCI pattern and clotting factor level).
ROC analysis was used to determine a threshold for discerning the skewing pattern.
The Wilcoxon rank-sum test was used to assess differences between the median coagulant factor values in different groups.
All P values were two-sided, with <0.05 considered statistically significant. All analyses were performed using the statistical software R, release 3.5.1 (R Foundation for Statistical Computing, Vienna, Austria)
Results
Fifty-six carriers of haemophilia A and seventeen carriers of haemophilia B showed low clotting FVIII and FIX activity with a median of 38 IU/dL (IQR:30–44) and 24 IU/dL (IQR:18–37), respectively (Table 1). A very low FVIII:C and FIX:C level (≤20 IU/dL) was observed in ten carriers of haemophilia A and five of haemophilia B. Table S1 (Supplementary material) shows in detail the FVIII and FIX coagulant plasma levels for each carrier of haemophilia A and B.
In almost all women with low FVIII:C level, VWF:Ag, VWF:RCo and FVIII binding to VWF were normal except for one who turned out to be a carrier of both severe haemophilia and von Willebrand disease type 1.
Genetic analysis of the F8 and F9 identified a heterozygous gene variant in 71 out of 73 symptomatic women (97%).
Different types of F8 genetic variants were detected, including intron 22 inversions (29%), large deletions (5%), small deletions/insertions (20%), non-sense variants (16%), splice site variants (4%), and missense variants (23%), as reported in Tables 1 and S1 (Supplementary material). No F8 gene variant was identified in two carriers of haemophilia A (3%) (Table 1).
Gene defects observed in 17 carriers of haemophilia B were mainly missense variants (53%), followed by nonsense variants (23%), small deletions (12%) and splice site variants (12%) (Tables 1 and S1, Supplementary material).
Phenotypic and genotypic data were anonymously submitted to the ClinVar database (ClinVar accession numbers: SCV001424858–SCV001424904) (Tables 1 and S1, Supplementary material).
Analysis and distribution of skewed XCI pattern
Three female carriers of haemophilia and 11 female non-carriers were uninformative due to HUMARA homozygosity and were thus excluded from the analysis.
Approximately 20% (14/70) of haemophilia carriers showed an extremely skewed XCI (≥90:10), while 33% (23/70) showed a skewed pattern of ≥80:20 (Table 2). In contrast, only 9% (6/68) and 14% (10/68) of the women in the group of non-carriers showed an extremely skewed or skewed XCI, respectively, similarly to the frequencies found in a control group (3.6% and 14.2%) reported in a previous study (Table 2) [10]. A skewed XCI (≥70:30) was detected in 57% (40/70) of haemophilia carriers in contrast to 26% (18/68) in the non-carriers group and 25% in a control group of women (Table 2) [10].
It was possible to distinguish mutant X chromosome and wild‐type X chromosome alleles in 13 cases with family history of haemophilia. In these cases, the mutant allele (F8 or F9), inherited from haemophilic parents, was predominantly active. In two cases, with no family history of haemophilia, the active X chromosome was the mutant F8 allele inherited from the mother, who was a carrier as confirmed by genetic testing (Table S1, Supplementary material).
Correlation between coagulant factor level and XCI pattern
To assess whether the low FVIII and FIX activity (≤50 IU/dL) in female carriers of haemophilia could be related to the degree of XCI pattern, we investigated the overall correlation between percentage of XCI pattern and coagulant factor levels of each carrier of haemophilia. A correlation between coagulant factor level and percentage of XCI pattern was found in haemophilia A and B carriers, with a value of r = −0.60, as shown in Fig. 1. Moreover, the same analysis was performed after separating cases of haemophilia A from haemophilia B. A Pearson’s value of r = −0.58 was found for carriers of haemophilia A and of r = −0.69 for carriers of haemophilia B, indicating that there was still a correlation between coagulant factor level and degree of XCI pattern even considering women with haemophilia A and B separately.

Dashed horizontal line indicates clotting factor level at 20 IU/dL and dashed vertical lines indicate XIC patterns 50%, 80% and 90%. Correlation between XIP and coagulant factor level was demonstrated using the Pearson method (r = −0.60). Black circles indicate female carriers of haemophilia A and grey circles female carriers of haemophilia B.
In particular, the group of carriers with coagulant factor level below 20 IU/dL showed a high degree of XCI pattern (≥80:20), which seems to be an appreciable determinant of low clotting factor level (Fig. 1). A statistical association between XCI pattern and level of FVIII:C and FIX:C was observed (p < 0.001). Ninety-three percent of haemophilia carriers with a clotting factor level of ≤20 IU/dL showed an XCI pattern of ≥80:20 (Table 3).
ABO group
ABO blood group (O or non-O) information was gathered for all haemophilia A carriers recruited in the study: 29 out of 56 with group O and 27 with group non-O. We found no significant difference in FVIII coagulant activity between group O [FVIII:C median 38 IU/dL (IQR: 30–44)] and non-O [FVIII:C median 37 IU/dL (IQR: 29–45)] (p = 0.82), suggesting that ABO locus alone was not sufficient to explain the variation observed in these carrier women with low FVIII levels (Table 1).
Bleeding symptoms and coagulant factor levels
In our cohort of haemophilia carriers, the most common bleeding symptoms were menorrhagia (74%) and cutaneous bleeding (69%). Twenty-six women out of 46 (54%) experienced bleeding after tooth extraction, while 20 out of 48 had bleeding after surgery (42%). Epistaxis was experienced in 43% of the cohort. Haemarthrosis and muscle haematomas, which are the most frequent symptoms in haemophilic patients, were reported only in 11 and 17%, respectively. Female carriers with these types of bleeds had lower factor coagulant activity than other carriers without these symptoms. The median factor level in carriers with haemarthrosis was 4 IU/dL (IQR: 3–7) versus 37 IU/dL (IQR: 28–44) in female carriers without haemarthrosis (p < 0.001); while in female carriers with muscle hematomas, the median factor coagulant level was 14 IU/dL (IQR:7–28) versus 38 IU/dL (IQR: 29–44) in carriers without haematomas (p < 0.001).
Female carriers with a BAT score ≥6 showed a median factor coagulant level of 28 IU/dL (IQR: 12–37), while haemophilia carriers with a BAT score <6 had a median factor coagulant level of 39 IU/dL (IQR: 30–44) (p = 0.003).
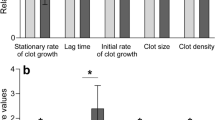
A comparative analysis of the BAT scores of two groups of haemophilia carriers was performed by dividing our cohort of carriers into two groups based on FVIII/FIX level and XCI pattern degree. Group A included female carriers with a coagulant factor level ≤20 IU/dL and skewed XCI pattern (≥80:20), while group B consisted of female carriers with a coagulant factor level >20 IU/dL and a random XCI pattern (<80:20). This analysis emphasized that group B had a median BAT score of 15 (IQR: 8–18), while female carriers belonging to group A showed a median BAT score of 5 (IQR: 2–8) (p = 0.001) (Fig. 2). In addition, we evaluated the relationship between the BAT scores of the two groups of women with haemophilia as a categorical variable, i.e. abnormal (≥6) as opposed to normal (<6). An abnormal BAT score (≥6) was found in 12 out of 14 carriers in group B. In group A, 16 women showed an abnormal BAT score, while 23 were normal. Therefore, the probability of having an abnormal BAT score in group B was 86% versus 41% for group A (p = 0.004).

Group A: carriers with factor level >20 IU/dL and XIC pattern <80:20. Group B: carriers with factor level ≤20 IU/dL and XIC pattern ≥80:20. Dashed horizontal line indicates the abnormal value of ISTH BAT score (≥6).
Gene defect and bleeding phenotype
To assess whether there was a relationship between the type of gene defect, bleeding phenotype and factor level, gene variants were clustered into null variants (intron 22 inversions, large deletions, small deletions/insertions, non-sense variants) and non-null variants (missenses and splice site variants) [29]. Female carriers of null variants with a BAT score ≥6 showed a median factor level of 34 IU/dL (IQR: 20–40), while female carriers of non-null variants with a BAT score ≥6 had a median factor level of 13 IU/dL (IQR: 10–23) (p = 0.053). Female carriers of null variants with a BAT score <6 had a median factor level of 39 IU/dL (IQR: 30–44), while carriers of non-null variants with a BAT score <6 had a median factor level of 42 IU/dL (IQR: 34–45) (p = 0.550).
Discussion
X-linked disorders are generally expressed in males while in female carriers the phenotype is more variable. Carriers, who inherit the mutated F8 or F9 gene of haemophilia, are expected to have ~50% of the normal population’s clotting factor levels due to X chromosome inactivation. The variability of phenotypic expression of the disease might be attributable to several genetic mechanisms, including the skewed silencing of the X chromosome. In this study, we analyzed the XIC pattern in symptomatic female carriers of haemophilia A and B, two X-linked disorders.
In our cohort, a specific, heterozygous single gene variant was found in 97% of female carriers, which alone did not justify their low factor levels. HUMARA assay results showed that low coagulant factor activity was associated with an unfavourable XCI skewing. In particular, a negative correlation was found in the carrier group between skewed XCI pattern (≥80:20) and very low coagulant factor level (≤20 IU/dL), as previously observed by Renault and colleagues [11]. It is worth noting that another study did not find a correlation between XCI and coagulant factor level in a cohort of haemophilia carriers [30]. Additionally, HUMARA locus analysis showed that 33% of haemophilia carriers had a skewed XCI of ≥80:20, while an extremely skewed XCI (≥90:10) was observed in 20% of female carriers. Previous estimates of the degree of XCI in other X-linked diseases, such as X-linked mental retardation and Lesch-Nyhan disease, report XCI values comparable with our data [31]. Conversely, an extremely skewed inactivation pattern is very rare in the normal population, with XCI ≥ 90:10 and XCI ≥ 80:20 in 3.6% and 14.2% of women in a control group, respectively [10, 32]. These data are similar to those found in our control group of non-carrier Italian females.
The high degree of XCI in women with X-linked disorders has been explained by recent data showing that a secondary cell selection occurs after an initially X-random inactivation process. It has been recognised that cell selection can occur both against those cells in which the gene variant is on the active X chromosome and against those cells in which the gene variant is on the inactive X chromosome [33]. Most of the time, the presence of an X-linked variant results in a growth disadvantage of cells expressing the mutated allele [33, 34], but exceptions have been reported [35,36,37].
In recent years, understanding of the X inactivation process has increased significantly, and it is believed that XCI is not only a biological process occurring early in embryonic life at the blastocyst stage, but also an important modifier of X-linked disorders in females. In humans, the timing of XCI choice and whether the choice occurs completely randomly or under a genetic influence is debated [38, 39].
The opinion that X-linked disorders are only relevant for males is becoming outdated, as emphasized by the large number of studies linking skewed X chromosome inactivation to the phenotype in female carriers with X-linked disorders such as haemophilia. Our data reveal that XCI seems to be an appreciable determinant of clotting factor residual level, with potential clinical significance particularly in haemophilia carriers in whom the genetic defect on a single allele cannot explain a very low factor level. XCI can influence the coagulant factor level and severity of haemophilia A in heterozygous female carriers, but a further contributing factor, such as ABO group, may also affect the factor level. Phenotypically normal individuals with blood group O could have a 25% reduction of FVIII coagulant activity compared to non-O individuals, due to the effects of the ABO locus on von Willebrand factor antigen [40, 41].
In our study, we found no correlation between blood group O versus non-O and coagulant FVIII levels, in agreement with previous findings [42, 43]. This could suggest that ABO locus alone is not sufficient to explain variation in FVIII coagulant level, although this finding may also be due to the small number of carriers analyzed.
The bleeding symptoms of men with haemophilia are well known, while little information is available on bleedings in haemophilia carriers. Historically, carriers were thought to have a limited bleeding phenotype, as the majority of them express about half the normal plasma factor level that is considered adequate for haemostasis [3]. Mounting evidence supports the notion that haemophilia carriers may exhibit an increased bleeding tendency despite coagulant factor levels within the normal laboratory range (greater than 2 standard deviations below the population mean) [44, 45]. An early publication in 1951 reported excessive bleeding after tooth extraction in 47% of haemophilia carriers [46]. The first description of a severe phenotype in a small cohort of female carriers dates to 1978 [47]. Subsequent publications consisted of a small cohort case series relying on non-standardized approaches to evaluate bleeding symptoms. These studies showed that the major episodes were prolonged bleeding after minor surgeries (tonsillectomy and tooth extractions), joint bleeds, or post-partum bleeding [48,49,50].
In 2006, a large cohort cross-sectional study examined the risk of bleeding in 225 carriers of haemophilia A and B compared with a group of 143 non-carriers [3]. An association among low clotting factor levels (≤40 IU/dL) and an increased risk of bleeding from small wounds and joint bleeds was highlighted. Subsequently, a multicenter study in the United States of women with FVIII and FIX levels between ≤1 IU/dL and 50 IU/dL showed that mucocutaneous bleeding was the most prevalent symptoms in this cohort of haemophilia carriers, while joint bleeds were also detected [51, 52]. Menorrhagia and post-partum bleeding were unexpectedly less frequent than reported prior to that study.
In our cohort of haemophilia carriers, the most frequent hemorrhagic symptoms were menorrhagia, cutaneous bleeding, prolonged bleeding after tooth extraction and epistaxis, in agreement with data from previous studies [17, 44, 52, 53]. Interestingly, 11 and 17% of women reported haemartrosis and muscle haematomas, respectively, which are the most severe types of bleeding in haemophilia patients, and a correlation was found between these types of bleeding and lower levels of the coagulant factor. This finding also emerged in a multinational study conducted by members of the Global Emerging Hemostasis Panel (GEHEP) on a large number of haemophilia carriers [17]. Haemarthrosis and muscle hematoma were reported in 9 and 11% of carriers, respectively, and were more frequent in those with low FVIII and FIX levels.
Additionally, in our cohort, when evaluating bleeding symptoms considering both severity and frequency according to BAT score, a significant difference in factor levels between the group of carriers with a high bleeding score (BAT ≥ 6) and the group with a low bleeding score (BAT < 6) was found, in agreement with previously reported data from a multinational study of haemophilia carriers [53]. The probability of having an abnormal BAT score in the group of women with factor activity ≤20 IU/dL and skewed XCI (≥80:20) was 86%, compared with 41% for the group with opposite conditions (factor activity >20 IU/dL and random XCI). Clinical data, collected from haemophilia carriers in our study, showed that even women who were mildly affected (≤20IU/dL), experienced severe symptoms including haemarthrosis and muscle bleeding.
Clinical data also showed a high prevalence of bleeding symptoms in haemophilia carriers, with many specific female symptoms (obstetrical and gynecological) and demonstrated that many carriers had low factor activity levels in the range of mild haemophilia as defined in males.
Low coagulant levels, associated with a skewed X chromosome inactivation, may explain the severe clinical evidence observed in our cohort of haemophilia carriers.
Moreover, carriers of non-null variants showed lower factor level and more severe clinical phenotype, though this is not statistically significant (p = 0.053). Further studies are needed to better elucidate mechanisms that connect bleeding phenotype and the type of underlying gene variant.
In conclusion, our study demonstrates that XCI might greatly influence the phenotype and expression of X-linked diseases, although it is only one of the factors contributing to the complexity of the haemophilia carrier phenotype.
References
Mannucci PM, Tuddenham EG. The Hemophilias–from royal genes to gene therapy. N Engl J Med. 2001;344:1773–9.
Peyvandi F, Garagiola I, Young G. The past and future of hemophilia: diagnosis, treatments, and its complications. Lancet. 2016;388:187–97.
Plug I, Mauser-Bunschoten EP, Bröcker-Vriends AH, van Amstel HK, van der Bom JG, van Diemen-Homan JE, et al. Bleeding in carriers of hemophilia. Blood. 2006;108:52–6.
Radic CP, Rossetti LC, Abelleyro MM, Tetzlaff T, Candela M, Neme D, et al. Phenotype-genotype correlations in hemophilia A carriers are consistent with the binary role of the phase between F8 and X-chromosome inactivation. J Thromb Haemost. 2015;13:530–9.
Miyawaki Y, Suzuki A, Fujimori Y, Takagi A, Murate T, Suzuki N, et al. Severe hemophilia A in a Japanese female caused by an F8-intron 22 inversion associated with skewed X chromosome inactivation. Int J Hematol. 2010;92:405–8.
Connallon T, Clark AG. Sex-differential selection and the evolution of X inactivation strategies. PLoS Genet. 2013;94:e1003440.
Orstavik KH. X chromosome inactivation in clinical practice. Hum Genet. 2009;126:363–73.
Carrel L, Willard HF. X-inactivation profile reveals extensive variability in X-linked gene expression in females. Nature 2005;434:400–4.
Viggiano E, Picillo E, Ergoli M, Cirillo A, Del Gaudio S, Politano L. Skewed X‐chromosome inactivation plays a crucial role in the onset of symptoms in carriers of Becker muscular dystrophy. J Gene Med. 2017;19:e2952.
Amos-Landgraf JM, Cottle A, Plenge RM, Friez M, Schwartz CE, Longshore J, et al. X chromosome inactivation patterns of 1005 phenotypically unaffected females. Am J Hum Genet. 2006;79:493–9.
Renault NK, Dyack S, Dobson MJ, Costa T, Lam WL, Greer WL. Heritable skewed X-chromosome inactivation leads to haemophilia A expression in heterozygous females. Eur J Hum Genet. 2007;15:628–37.
Favier R, Lavergne JM, Costa JM, Caron C, Mazurier C, Viémont M, et al. Unbalanced X-chromosome inactivation with a novel FVIII gene mutation resulting in severe hemophilia A in a female. Blood 2000;9613:4373–75.
Boban A, Lambert C, Lannoy N, Hermans C. Comparative study of the prevalence of clotting factor deficiency in carriers of haemophilia A and haemophilia B. Haemophilia. 2017;23:e471–3.
Hermans C, Kulkarni R. Women with bleeding disorders. Haemophilia. 2018;24:29–36.
Srivastava A, Brewer AK, Mauser-Bunschoten EP, Key NS, Kitchen S, Llinas A, et al. Treatment guidelines working group on behalf of the world federation of hemophilia. Guidelines for the management of haemophilia. Haemophilia 2013;19:e1–47.
Potgieter JJ, Damgaard M, Hillarp A. One-stage vs. chromogenic assays in haemophilia A. Eur J Haematol. 2015;94:38–44.
James PD, Mahlangu J, Bidlingmaier C, Mingot-Castellano ME, Chitlur M, Fogarty PF, et al. Global emerging hemostasis experts panel (GEHEP). Evaluation of the utility of the ISTH-BAT in haemophilia carriers: a multinational study. Haemophilia. 2016;22:912–8.
Stufano F, Baronciani L, Mane-Padros D, Cozzi G, Faraudo S, Peyvandi F. A comparative evaluation of a new fully automated assay for von Willebrand factor collagen binding activity to an established method. Haemophilia. 2018;24:156–61.
Federici AB, Canciani MT, Forza I, Mannucci PM, Marchese P, Ware J, et al. A sensitive ristocetin co-factor activity assay with recombinant glycoprotein Ibalpha for the diagnosis of patients with low von Willebrand factor levels. Haematologica 2004;89:77–85.
Mazurier C, Gaucher C, Jorieux S, Parquet- Gernez A, Goudemand M. Evidence for a von Willebrand factor defect in factor VIII binding in three members of a family previously misdiagnosed mild haemophilia A and haemophilia A carriers: consequences for therapy and genetic counselling. Br J Haematol. 1990;76:372–9.
Miller I, Djkes DD. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acid Res. 1988;16:1215–20.
Liu Q, Nozari G, Sommer SS. Single-tube polymerase chain reaction for rapid diagnosis of the inversion hotspot of mutation in hemophilia A. Blood. 1998;92:1458–9.
Bagnall RD, Waseem N, Green PM, Giannelli F. Recurrent inversion breaking intron 1 of the factor VIII gene is a frequent cause of severe hemophilia A. Blood 2002;99:168–74.
Lee JC, Chang JG. ABO genotyping by polymerase chain reaction. J Forensic Sci. 1992;37:1269–75.
Ota M, Fukushima H, Kulski JK, Inoko H. Single nucleotide polymorphism detection by polymerase chain reaction-restriction fragment length polymorphism. Nat Protoc. 2007;2:2857–64.
Allen RC, Zoghbi HY, Moseley AB, Rosenblatt HM, Belmont JW. Methylation of HpaII and HhaI sites near the polymorphic CAG repeat in the human androgen-receptor gene correlates with X chromosome inactivation. Am J Hum Genet. 1992;51:1229–39.
Rodeghiero F, Tosetto A, Abshire T, Arnold DM, Coller B, James P, et al. ISTH/SSC joint VWF and Perinatal/Pediatric Hemostasis Subcommittees Working Group. ISTH/SSC bleeding assessment tool: a standardized questionnaire and a proposal for a new bleeding score for inherited bleeding disorders. J Thromb Haemost. 2010;8:2063–5.
Elbatarny M, Mollah S, Grabell J, Bae S, Deforest M, Tuttle A, et al. Normal range of bleeding scores from the ISTH-BAT: adult and pediatric data from the merging project. Haemophilia. 2014;20:831–5.
Peyvandi F, Mannucci PM, Garagiola I, El‑Beshlawy, Elalfy M, Ramanan V, et al. Randomized trial of factor VIII and neutralizing antibodies in hemophilia A. N Engl J Med. 2016;374:2054–64.
Ørstavik KH, Scheibel E, Ingerslev J, Schwartz M. Absence of correlation between X chromosome inactivation pattern and plasma concentration of factor VIII and factor IX in carriers of haemophilia A and B. Thromb Haemost. 2000;83:433–7.
Torres RJ, Puig JG. Skewed X inactivation in Lesh-Nyhan disease carrier females. J Hum Genet. 2017;62:1079–83.
Bolduc V, Chagnon P, Provost S, Dubé MP, Belisle C, Gingras M, et al. No evidence that skewing of X chromosome inactivation patterns is transmitted to offspring in humans. J Clin Invest. 2008;118:333–41.
Ørstavik KH. Skewed X inactivation in healthy individuals and in different diseases. Acta Paediatr Suppl. 2006;95:24–9.
Lacout C, Haddad E, Sabri S, Svinarchouk F, Garçon L, Capron C, et al. A defect in hematopoietic stem cell migration explains the nonrandom X-chromosome inactivation in carriers of Wiskott-Aldrich syndrome. Blood 2003;102:1282–9.
Migeon BR, Moser HW, Moser AB, Axelman J, Sillence D, Norum RA. Adrenoleukodystrophy: evidence for X linkage, inactivation, and selection favoring the mutant allele in heterozygous cells. Proc Natl Acad Sci USA. 1981;78:5066–70.
Maier EM, Kammerer S, Muntau AC, Wichers M, Braun A, Roscher AA. Symptoms in carriers of adrenoleukodystrophy relate to skewed X inactivation. Ann Neurol. 2002;52:683–8.
Bicocchi MP, Migeon BR, Pasino M, Lanza T, Bottini F, Boeri E, et al. Familial nonrandom inactivation linked to the X inactivation centre in heterozygotes manifesting haemophilia A. Eur J Hum Genet. 2005;13:635–40.
Renault NK, Pritchett SM, Howell RE, Greer WL, Sapienza C, Ørstavik KH, et al. Human X-chromosome inactivation pattern distributions fit a model of genetically influenced choice better than models of completely random choice. Eur J Hum Genet. 2013;21:1396–402.
Orstavik KH, Orstavik RE, Schwartz M. Skewed X chromosome inactivation in a female with haemophilia B and in her non-carrier daughter: a genetic influence on X chromosome inactivation? J Med Genet. 1999;36:865–6.
O’Donnell J, Boulton FE, Manning RA, Laffan MA. Genotype at the secret or blood group locus is a determinant of plasma von Willebrand factor level. Br J Haematol. 2002;116:350–6.
Sousa NC, Anicchino-Bizzacchi JM, Locatelli MF, Castro V, Barjas-Castro ML. The relationship between ABO groups and subgroups, factor VIII and von Willebrand factor. Haematologica. 2007;92:236–9.
Loomans JI, van Velzen AS, Eckhardt CL, Peters M, Mäkipernaa A, Holmstrom M, et al. Variation in baseline factor VIII concentration in a retrospective cohort of mild/moderate hemophilia A patients carrying identical F8 mutations. J Thromb Haemost. 2017;15:246–54.
Ay C, Thom K, Abu-Hamdeh F, Horvath B, Quehenberger P, Male C, et al. Determinants of factor VIII plasma levels in carriers of haemophilia A and in control women. Haemophilia. 2010;16:111–7.
Paroskie A, Gailani D, DeBaun MR, Sidonio RF Jr. A cross-sectional study of bleeding phenotype in haemophilia A carriers. Br J Haematol. 2015;170:223–8.
Olsson A, Hellgren M, Berntorp E, Ljung R, Baghaei F. Clotting factor level is not a good predictor of bleeding in carriers of haemophilia A and B. Blood Coagul Fibrinolysis. 2014;25:471–5.
Merskey C, Macfarlane RG. The female carrier of haemophilia. A clinical and laboratory study. Lancet. 1951;260:487–90.
Lusher LM, McMillan CW. Severe factor VIII and factor IX deficiency in females. Am J Med. 1978;65:637–48.
Mauser-Bunschoten EP, van Houwelingen JC, Sjamsoedin-Visser EJ, van Dijken PJ, Kok AJ, Sixma JJ. Bleeding symptoms in carriers of haemophilia A and B. Thromb Haemost. 1988;59:349–52.
Wahlberg T. Carriers and noncarriers of haemophilia A. Evaluation of bleeding symptoms registered by a self-administered questionnaire with binary (no/yes) questions. Thromb Res. 1982;25:415–22.
Yang MY, Ragni MV. Clinical manifestations and management of labor and delivery in women with factor IX deficiency. Haemophilia. 2004;10:483–90.
Di Michele DM, Gibb C, Lefkowitz JM, Ni Q, Gerber LM, Ganguly A. Severe and moderate haemophilia A and B in US females. Haemophilia. 2014;20:e136–43.
Paroskie A, Oso O, Almassi B, DeBaun MR, Sidonio RF Jr. Both hemophilia health care providers and hemophilia a carriers report that carriers have excessive bleeding. J Pediatr Hematol Oncol. 2014;36:e224–30.
Raso S, Lambert C, Boban A, Napolitano M, Siragusa S, Hermans C. Can we compare haemophilia carriers with clotting factor deficiency to male patients with mild haemophilia? Haemophilia. 2020;26:117–21.
Acknowledgements
The authors thank Dr Luigi Flaminio Ghilardini for help with the figures.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
ES is a member of advisory committees (Sobi, NovoNordisk, Roche, Shire, Bayer, Kedrion, Grifols, Pfizer) and speaker bureaus (Octapharma, Kedrion, Grifols, Bayer, Roche, Shire, Bioverativ, Sobi, NovoNordisk, Pfizer, CSL Behring). FP has received honoraria for participating as a speaker at satellite symposia organised by Bioverativ, Grifols, Roche, Sanofi, Sobi, Spark and Takeda. FP reports participation on the advisory boards of Sanofi and Sobi.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Garagiola, I., Mortarino, M., Siboni, S.M. et al. X Chromosome inactivation: a modifier of factor VIII and IX plasma levels and bleeding phenotype in Haemophilia carriers. Eur J Hum Genet 29, 241–249 (2021). https://doi.org/10.1038/s41431-020-00742-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-020-00742-4
This article is cited by
-
In vivo LNP-CRISPR Approaches for the Treatment of Hemophilia
Molecular Diagnosis & Therapy (2024)
-
Clinical Analysis and Mental Health Survey of Hemophilia Carriers: a Cross-sectional Study
Current Medical Science (2024)
-
A novel quantitative targeted analysis of X-chromosome inactivation (XCI) using nanopore sequencing
Scientific Reports (2023)
-
Yield of clinically reportable genetic variants in unselected cerebral palsy by whole genome sequencing
npj Genomic Medicine (2021)
