
Abstract
In about 30% of infantile, juvenile, or adolescent patients with steroid-resistant nephrotic syndrome (SRNS), a monogenic cause can be identified. The histological finding in SRNS is often focal segmental glomerulosclerosis (FSGS). Genetic data on adult patients are scarce with low diagnostic yields. Exome sequencing (ES) was performed in patients with adult disease onset and a high likelihood for hereditary FSGS. A high likelihood was defined if at least one of the following criteria was present: absence of a secondary cause, ≤25 years of age at initial manifestation, kidney biopsy with suspicion of a hereditary cause, extrarenal manifestations, and/or positive familial history/reported consanguinity. Patients were excluded if age at disease onset was <18 years. In 7/24 index patients with adult disease onset, a disease-causing variant could be identified by ES leading to a diagnostic yield of 29%. Eight different variants were identified in six known genes associated with monogenic kidney diseases. Six of these variants had been described before as disease-causing. In patients with a disease-causing variant, the median age at disease onset and end-stage renal disease was 26 and 38 years, respectively. The overall median time to a definite genetic diagnosis was 9 years. In 29% of patients with adult disease onset and suspected hereditary FSGS, a monogenic cause could be identified. The long delay up to the definite genetic diagnosis highlights the importance of obtaining an early genetic diagnosis to allow for personalized treatment options including weaning of immunosuppressive treatment, avoidance of repeated renal biopsy, and provision of accurate genetic counseling.
Similar content being viewed by others


Introduction
The advances made possible by next-generation sequencing (NGS) techniques have shown that in about 30% of infantile, juvenile, or adolescent patients with steroid-resistant nephrotic syndrome (SRNS), a monogenic cause can be identified [1]. The histological correlate in patients with SRNS is often focal segmental glomerulosclerosis (FSGS) or minimal change disease (MCD) in early stages [2]. So far, more than 50 genes have been associated with hereditary FSGS/SRNS [3,4,5,6,7]. The clinical manifestation of hereditary FSGS is extremely variable with differences in expressivity, apparent by different ages of disease onset, progression to end-stage renal disease (ESRD) or the presence of nephrotic syndrome (NS). NS is defined by significant proteinuria (>40 mg/m2 per hour). Patients with NS typically present with hypoalbuminemia causing severe edema. Progression of chronic kidney disease gradually leads to ESRD requiring dialysis or transplantation. Most importantly, this has a strong impact on patient survival. In contrast, in adult patients with FSGS, SRNS, or NS, hereditary causes are widely neglected differential diagnoses.
Genetic studies have assigned a pivotal role to the podocyte in terms of pathophysiology of hereditary FSGS/SRNS. Multiple genes encoding slit membrane components, actin binding proteins, proteins important for coenzyme Q10 biosynthesis, as well as nuclear transcription factors have been associated with monogenic-caused podocyte dysfunction [5]. These genes are highly expressed in the podocyte leading to their degeneration with consecutive symptoms in the presence of disease-causing variants [likely pathogenic or pathogenic variants as per the American College of Human Genetics (ACMG) with a fitting genotype]. Furthermore, variants in the COL4A3-5 genes, encoding elements of the glomerular basement membrane and associated with Alport syndrome, can mimic the histological and/or clinical picture of FSGS/SRNS [8, 9]. Most literature is limited due to the use of panel diagnostics or targeted single-gene sequencing. The heterogeneous presentation in hereditary FSGS/SRNS favors a hypothesis-free approach like exome or genome sequencing (ES or GS) to adequately address its genetically heterogenous nature.
It is assumed that in at least 10% of adults who require renal replacement therapy a hereditary cause can be identified. This would represent one of the most common causes of ESRD next to diabetes, hypertension, glomerulonephritis, and pyelonephritis [10]. The likelihood for identifying a disease-causing variant resulting in FSGS/SRNS is inversely related to age of onset due to usually early manifestation in monogenic cases [1, 5, 11, 12]. The diagnostic yield in patients above 21 years at study entry with glomerulopathies ranges from 7% in unselected cohorts [13] to 12–14% in selected cohorts [14, 15]. Overall, genetic and phenotypic data on patients with FSGS or SRNS disease onset above 18 years are scarce. Only few monogenic causes have been identified in adult patients, with a mean FSGS disease onset ranging from 26 years in patients with disease-causing variants in classical FSGS genes to 36 years in patients with disease-causing variants in COL4A3-5 [16].
Using ES in a cohort of 24 adult patients with suspected hereditary FSGS/SRNS, we aimed to identify disease-causing variants.
Material and methods
Study population
The study was approved by the local Ethics Committee of the Technical University of Munich and performed according to standards of the 2013 Helsinki Declaration. Written informed consent was obtained from patients or their legal guardians.
For inclusion, a selection process was applied for adult disease onset patients with assumed hereditary FSGS or MCD to increase the likelihood for the identification of a monogenic cause. At least one of the following criteria had to be met to perform ES: absence of a secondary cause for FSGS, ≤25 years of age at initial manifestation, kidney biopsy with suspicion of a hereditary cause (i.e., ultrastructural changes of the glomerular basement membrane in electron microscopy [17]), extrarenal manifestations (syndromal disease, for example intellectual disability, eye or skeletal abnormalities), and/or positive familial history/reported consanguinity. Exclusion criteria were missing written informed consent, age below 18 years at initial manifestation of kidney disease, and absence of the above-mentioned inclusion criteria.
To identify patients with a high likelihood for a hereditary cause, we performed a screening of 1700 adult kidney disease patients treated at our university hospital between January 2000 and February 2018. In addition, we received external samples meeting the inclusion criteria (Fig. 1). Origin was classified into European (non-Finnish) and other origins.

A total of 1700 in-house individuals were screened for suspected hereditary FSGS. Decision to include patients was based on prioritization criteria, which led to the inclusion of 19 index patients. In addition, five samples were sent in for molecular analysis. A total of 29% of the cases could be solved. A total of 68% of the patients did not show any clear disease-causing variant.
Clinical case information
Clinical and phenotype information was retrieved from clinical reports and medical history. Age of disease onset was defined as the earliest report of a kidney disease in the medical history (e.g., first onset of proteinuria, edema, hospitalization due to a kidney disease, etc.). Age of onset of ESRD was determined as the beginning of renal replacement therapy (hemodialysis or peritoneal dialysis) or preemptive kidney transplantation. A standardized questionnaire was used for the assessment of clinical information. Stage of chronic kidney disease was assessed at the latest available date according to KDIGO guidelines [18].
Genetics
DNA was extracted from peripheral blood using the Gentra Puregene Blood Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. ES was performed using a Sure Select Human All Exon 60 Mb V6 Kit (Agilent) and a HiSeq4000 (Illumina) or a Sure Select Human All Exon 60 Mb V5 Kit (Agilent) and a HiSeq2500 (Illumina) as previously described [19]. Mitochondrial DNA was derived from off-target exome reads as previously described [20]. Reads were aligned to the human reference genome (UCSC Genome Browser build hg19) using Burrows-Wheeler Aligner (v.0.7.5a). Detection of single-nucleotide variants and small insertions and deletions (indels) was performed with SAMtools (version 0.1.19). Exome depth was used for the detection of copy-number variations [21]. Confirmation of the identified variants and segregation analysis was carried out by Sanger sequencing. For the analysis of de novo, autosomal dominant and mitochondrial variants, only variants with a minor allele frequency of less than 0.1% in the in-house database of the Helmholtz center Munich consisting of over 16,000 exomes were considered. For the analysis of autosomal recessive and X-linked variants [homozygous, hemizygous, or (putatively) compound heterozygous], only variants with a minor allele frequency of less than 1% were considered. For the NPHS2 p.(Arg229Gln) allele a MAF > 1% was accepted due to the known association with hereditary nephropathy when found in trans with a (likely) pathogenic variant [22, 23]. Furthermore, variants were compared to publicly available databases such as the Genome Aggregation Database (gnomAD). Only variants rated as “likely pathogenic” or “pathogenic” according ACMG classification and recent amendments and with a genotype in agreement with the mode of inheritance led to the designation “solved case” [24, 25]. To confirm if a variant was already published, a data search for the respective variant was performed in PubMed, ClinVar, and the Human Gene Mutation Database.
In addition, in one case (F332) with positive familial history and suspected autosomal dominant inheritance, MUC1 analysis was performed as previously described by Southern blot after unremarkable ES [26, 27].
Identified variants were submitted to the Leiden Open Variation Database (LOVD; https://databases.lovd.nl/shared) and to ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/).
Statistics
Categorical data are presented as absolute and relative frequencies. Continuous variables are expressed as median and interquartile range (IQR, 25th–75th percentile). For the calculation of the diagnostic yield only pathogenic and likely pathogenic variants with a fitting genotype (disease-causing variants) according to the definition of the ACMG standards and guidelines (i.e., classified as “solved cases”) were used. To test for group differences t-test was used and presented as mean ± standard deviation. To examine disease and ESRD onset as well as duration until the definite genetic diagnosis only “solved cases” were considered. All statistical tests were two-sided and p values < 0.05 were considered significant. Statistical analysis was performed using SPSS version 23.0 (SPSS Inc., Chicago, IL, USA).
Results
A total of 1700 in-house patients were screened and 19 index patients were included into the study. These index patients came from 19 unrelated families comprising a total of 44 individuals. Furthermore, five index patients from unrelated families comprising six individuals were submitted from external cooperation partners (Fig. 1).
Overall, 37 (74%) of the total 50 individuals were affected. Overall, 26 (70%) of these 37 affected individuals were male and 11 (30%) were female. Reported consanguinity was present in 2 (8%) families. Overall, 20 (83%) families were of European (non-Finnish) origin.
Of the overall 24 index patients, trio ES was done in three index cases (13%), duo ES in two (8%), and singleton ES in 19 (79%). In seven index patients, a disease-causing variant could be identified, corresponding to a total diagnostic yield of 29%. Overall, 5/7 index patients with a disease-causing variant were internal samples (diagnostic yield of the internal samples: 26%). Overall, 17 (71%) index patients could not be solved genetically.
Genetic variants
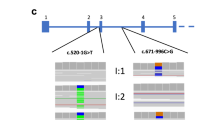
Overall, eight different variants were present in six known disease-causing genes (Table 1). Six of these variants have been described before as disease-causing. Overall, 2 (22%) variants, so far not described before, could be classified as disease-causing (likely pathogenic and pathogenic) according to ACMG guidelines (Table 1): the variant in COL4A3, c.2126-1G>C, p.(?) was present in a compound-heterozygous state with the variant c.4421T>C, p.(Leu1474Pro) and absent in gnomAD. The heterozygous variant in COL4A5, c.2359G>A, p.(Gly787Arg), was absent in our in-house database as well as in gnomAD. All identified variants reported in this study were at least conserved until M. musculus. In unsolved cases, no disease-causing variants could be detected in known disease-associated genes and no variants in candidate genes could be prioritized.
Age of disease and ESRD onset, and time to definite genetic diagnosis
The median age of disease onset and of ESRD of all 37 affected individuals was 26.0 [19.4–32.3] and 38.0 [28.4–39.6] years, respectively. In patients with a disease-causing variant, median age of disease onset and of ESRD was 25.0 [18.5–32.0] and 38.0 [24.0–39.0] years, respectively. Independent t-test showed that age of disease onset was not significantly different in solved versus unsolved cases (25.1 ± 5.9 vs 31.5 ± 14.3 years; p = 0.09). Similarly, age at ESRD did not differ significantly between solved and unsolved cases (32.6 ± 8.8 vs 44.9 ± 16.4 years; p = 0.15).
Overall, 15 (63%) out of 24 index patients had a disease onset ≥ 25 years. Of these 15 patients with disease onset ≥ 25 years, in 4 (27%) patients, a disease-causing variant was detected, and in 11 (73%) patients, no disease-causing variant could be identified.
Looking at the overall time to diagnosis in solved cases from the first manifestation of a renal disease to the definite genetic diagnosis the median time was 9.0 [2.9–19.0] years, with a maximum of 37 years and a minimum of 0 years (i.e., predictive molecular analysis in several family members of F103).
Clinical presentation and biopsy result
Overall, in all 37 affected individuals, the most frequent initial clinical presentation was proteinuria in 16 (44%) patients, followed by decline in kidney function in 7 (19%), edema in 4 (11%), and arterial hypertension in 3 (8%). Further causes were NS (n = 2; 6%), flank pain/nephrolithiasis (n = 2; 6%), or hematuria alone or combined with proteinuria (n = 2; 6%). In one affected sibling no data was available (Fig. 2a).

a The initial clinical presentation of the affected individuals. b The kidney biopsy results available from 21 affected individuals. AS Alport syndrome, FSGS focal segmental glomerulosclerosis, GN glomerulonephritis, MCD minimal change disease.
Concerning the cases in which a genetic diagnosis was made, 9 (64%) patients presented with proteinuria, and 2 (14%) patients with decline in kidney function, followed by NS (n = 1; 7%), arterial hypertension (n = 1; 7%), and edema (n = 1; 7%).
From the overall sample of all affected individuals, in 21 (57%) patients a kidney biopsy was available. Most frequent histological results showed FSGS in 12 (57%) patients or MCD in 4 (19%) patients. Other findings were global glomerulosclerosis (n = 2; 10%), lesions compatible with AS (n = 1; 5%), glomerulonephritis with mesangioproliferation (n = 1; 5%), and postinfectious glomerulonephritis (n = 1; 5%) (Fig. 2b).
Discussion
We were able to identify a disease-causing variant by ES in 29% (7/24) of adult patients with clinically suspected hereditary FSGS. This diagnostic yield is comparable to studies performed in children or adolescent patients [1]; however, it is so far unparalleled in adult patients. Compared to cohorts with a similar age structure, our diagnostic yield of 29% in patients with disease onset ≥18 years is quite high [14,15,16]. This high diagnostic yield could be explained by several factors.
Adult patients were included based on specific criteria. This selection process allowed us to include patients with disease onset ≥ 18 years of age while maintaining a potential high likelihood for the identification of a monogenic cause. Compared to the overall number of in-house screened patients, 19 included in-house index patients represent a stringently selected cohort. In all selected patients, comprehensive genetic testing by ES was performed, therefore, enabling the identification of variants throughout the exome instead of focusing on a limited set of genes via panel diagnostics. For future investigations, we plan on creating a scoring system based on the above-mentioned criteria for the identification of patients with a high likelihood of a hereditary nephropathy in adult age of onset.
Interestingly, in a cohort (n = 135) of only nonsyndromic FSGS and/or SRNS adult cases without family history, a quite high diagnostic yield of 12% has been reported [15]. This highlights the importance of hereditary causes in sporadic cases because of de novo variants. Trio ES was only performed in 13% of our cohort.
In addition to the high diagnostic yield in our adult patient cohort, we could identify two novel (likely) pathogenic variants in known disease-causing genes.
In our cohort, genetic heterogeneity is illustrated by three cases with a different genetic diagnosis than initially clinically presumed (FSGS-mimics/-phenocopies) (three out of seven solved cases; 43%). Two out of these three cases (F9 and F274) had disease-causing variants in the COL4A genes that have been frequently associated with hereditary FSGS [8, 9, 28]. Furthermore, one case with FGGS had a pathogenic variant in MUC1. Two of these three cases should be highlighted: the index patient, in whom a heterozygous disease-causing variant in COL4A5 was identified (F274), was female. She presented without the full picture of Alport syndrome but chronic renal failure with FSGS/FGGS on biopsy illustrating the wide range of phenotypes of X-linked Alport syndrome in females. Furthermore, we identified a cytosine insertion in the VNTR region of MUC1 (on additional targeted testing), which has been associated with ADTKD-MUC1; however, our patient (F332) presented with a creatinine increase and FGGS in the kidney biopsy, which has been performed late in the disease course, i.e., 3 years before preemptive living kidney donation. Although, in this case, F332, kidney biopsy showed FGGS rather than FSGS, the family was still included in this study as in the progress of the disease a more diffuse and global pattern of sclerosis can occur [29, 30]. Furthermore, there was a high likelihood of a hereditary kidney disorder as several individuals in the family were affected (Table 1). These two cases show that (i) the unbiased approach of ES can help to unravel cases with an ambiguous phenotype (F274), and (ii) limitations of ES should be known and additional methodologic approaches employed as, for example, the cytosine insertion in the VNTR region of MUC1 cannot be identified by ES (F332). The presence of FSGS-mimics/-phenocopies emphasizes that histologically diagnosed FSGS/FGGS can represent the final common track of several hereditary kidney disease entities. This is also underlined by a recently published case of a misclassification of ADTKD-UMOD as FSGS [31].
Clinical implications
The long period of 9 years until the final genetic diagnosis in patients with disease onset ≥ 18 years of age could be made highlights the importance of routinely including the possibility of a hereditary cause in the differential diagnosis of FSGS also in adult patients. Patients with a disease-causing variant reached ESRD earlier than unsolved cases; however, the difference was not significant. Larger studies need to evaluate if the presence of a disease-causing variant leads to earlier onset of ESRD. Furthermore, the long delay until a genetic diagnosis was made could be explained by three factors: (i) the presence of FSGS/SRNS-mimics/-phenocopies which makes it difficult to classify the clinical picture; (ii) the reluctant application of molecular techniques (e.g., NGS), which are still not widely used in clinical routine, at least in adult nephrology, and (iii) the missing awareness for hereditary causes of chronic kidney disease in adult patients.
In general, the histological term “FSGS” applied for primary (idiopathic, immunological), secondary, and hereditary causes represents a very broad disease spectrum [29]. As seen in our study, selection of patients is challenging especially in adult disease onset patients due to possible discrepancies of the histological description and the molecular genetic diagnosis. In some biopsies (see Table 1), the histological picture did not fit the molecular genetic diagnosis or was different in consecutive biopsies (for example, F103 and F27). This phenotypic variability further highlights the precise definition of an underlying genetic cause by molecular testing in patients with a clinically presumed hereditary disease. In our cohort, in genetically diagnosed cases, only one patient initially presented with NS typical for FSGS. The most common initial presentation was non-nephrotic proteinuria (64%). Therefore, NS associated with disease-causing variants in FSGS genes could be primarily present in cases with early childhood onset [32]. However, it has to be noted that only three of the six identified causative variants were located in classic FSGS genes. And, interestingly, case F9 had the genetic diagnose of Alport syndrome but the unusual adult manifestation of NS.
In terms of treatment and management the number of hereditary causes detected in children with SRNS is much higher than in adult patients [33]. In SRNS, the long period of time between the first disease manifestation and the detection of the definite genetic diagnosis increases the risk of unnecessary immunosuppressive treatment. This immunosuppressive treatment is known to be effective in a subset of patients with SRNS but patients with a hereditary cause do not benefit from it [34]. Furthermore, in families with an index case and confirmed genetic diagnosis, the selection of a relative as a kidney donor should be evaluated carefully because in some hereditary causes, disease up to ESRD can occur later in life [35].
Furthermore, there is a small cohort of monogenic disorders that can be influenced by treatment. These include patients with pathogenic variants in the genes encoding enzymes of the coenzyme Q10 biosynthesis [36,37,38]. In our cohort, two affected individuals of one consanguineous family had a disease-causing variant in COQ8B. Several case reports suggest a beneficial effect of coenzyme Q10 supplementation [38, 39].
In general, an early genetic diagnosis could allow a personalized treatment approach with weaning of immunosuppressive treatment, avoidance of renal biopsy, and provision of accurate genetic counseling [40]. Furthermore, genetic diseases can have multisystemic complications that need to be taken into consideration by the clinician, which is illustrated by F520 where the presence of Frasier syndrome is associated with an increased risk for gonadoblastoma [41].
Several limitations have to be mentioned. The number of affected individuals is rather small, therefore, a statement on the age of disease onset, ESRD, initial presentation as well as the kidney biopsy results is limited and not generalizable. In addition, patients in this study were included mainly in the way of a retrospective case selection. Therefore, data on the course of the immunosuppressive treatment as well as on steroid-resistance vs steroid-sensitivity were not readily available. Due to a pragmatic and time-efficient approach for scoring the 1700 in-house screened patients, data about the distribution of primary versus secondary FSGS were not systematically assessed. Furthermore, precise data concerning family history or consanguinity were not documented in most of the 1700 in-house screened patients, potentially leading to the relatively low number of only 1.1% included patients. Therefore, independent cohorts are needed to evaluate if the inclusion criteria used in this study do increase the likelihood for the presence of monogenic disease cause when selecting patients for molecular genetic testing.
Conclusion
We were able to identify a monogenic cause in more than one fourth of selected adult patients with suspected hereditary FSGS. This high diagnostic yield could only be achieved by including patients with a high likelihood for a hereditary disease cause based on specific selection criteria. The long time period until genetic diagnosis highlights the importance of comprehensive genetic testing, e.g., ES, to obtain an early genetic diagnosis that allows a personalized treatment approach with weaning of immunosuppressive treatment, avoidance of renal biopsy, and provision of accurate genetic counseling.
References
Sadowski CE, Lovric S, Ashraf S, Pabst WL, Gee HY, Kohl S, et al. A single-gene cause in 29.5% of cases of steroid-resistant nephrotic syndrome. J Am Soc Nephrol. 2015;26:1279–89.
Benoit G, Machuca E, Heidet L, Antignac C. Hereditary kidney diseases: highlighting the importance of classical Mendelian phenotypes. Ann N Y Acad Sci. 2010;1214:83–98.
Ashraf S, Kudo H, Rao J, Kikuchi A, Widmeier E, Lawson JA, et al. Mutations in six nephrosis genes delineate a pathogenic pathway amenable to treatment. Nat Commun. 2018;9:1960.
Lepori N, Zand L, Sethi S, Fernandez-Juarez G, Fervenza FC. Clinical and pathological phenotype of genetic causes of focal segmental glomerulosclerosis in adults. Clin Kidney J. 2018;11:179–90.
Lovric S, Ashraf S, Tan W, Hildebrandt F. Genetic testing in steroid-resistant nephrotic syndrome: when and how? Nephrol Dial Transpl. 2016;31:1802–13.
Varner JD, Chryst-Stangl M, Esezobor CI, Solarin A, Wu G, Lane B, et al. Genetic testing for steroid-resistant-nephrotic syndrome in an outbred population. Front Pediatrics. 2018;6:307.
Bierzynska A, McCarthy HJ, Soderquest K, Sen ES, Colby E, Ding WY, et al. Genomic and clinical profiling of a national nephrotic syndrome cohort advocates a precision medicine approach to disease management. Kidney Int. 2017;91:937–47.
Malone AF, Phelan PJ, Hall G, Cetincelik U, Homstad A, Alonso AS, et al. Rare hereditary COL4A3/COL4A4 variants may be mistaken for familial focal segmental glomerulosclerosis. Kidney Int. 2014;86:1253–9.
Braunisch MC, Büttner-Herold M, Günthner R, Satanovskij R, Riedhammer KM, Herr P-M, et al. Heterozygous COL4A3 variants in histologically diagnosed focal segmental glomerulosclerosis. Front Pediatrics. 2018;6:171.
Devuyst O, Knoers NV, Remuzzi G, Schaefer F. Rare inherited kidney diseases: challenges, opportunities, and perspectives. Lancet. 2014;383:1844–59.
Hinkes B, Vlangos C, Heeringa S, Mucha B, Gbadegesin R, Liu J, et al. Specific podocin mutations correlate with age of onset in steroid-resistant nephrotic syndrome. J Am Soc Nephrol. 2008;19:365–71.
Vivante A, Hildebrandt F. Exploring the genetic basis of early-onset chronic kidney disease. Nat Rev Nephrol. 2016;12:133–46.
Groopman EE, Marasa M, Cameron-Christie S, Petrovski S, Aggarwal VS, Milo-Rasouly H, et al. Diagnostic utility of exome sequencing for kidney disease. N Engl J Med. 2019;380:142–51.
Santin S, Bullich G, Tazon-Vega B, Garcia-Maset R, Gimenez I, Silva I, et al. Clinical utility of genetic testing in children and adults with steroid-resistant nephrotic syndrome. Clin J Am Soc Nephrol. 2011;6:1139–48.
Gribouval O, Boyer O, Hummel A, Dantal J, Martinez F, Sberro-Soussan R, et al. Identification of genetic causes for sporadic steroid-resistant nephrotic syndrome in adults. Kidney Int. 2018;94:1013–22.
Yao T, Udwan K, John R, Rana A, Haghighi A, Xu L, et al. Integration of genetic testing and pathology for the diagnosis of adults with FSGS. Clin J Am Soc Nephrol. 2019;14:213–23.
Haas M. Alport syndrome and thin glomerular basement membrane nephropathy: a practical approach to diagnosis. Arch Pathol Lab Med. 2009;133:224–32.
Levey AS, de Jong PE, Coresh J, El Nahas M, Astor BC, Matsushita K, et al. The definition, classification, and prognosis of chronic kidney disease: a KDIGO Controversies Conference report. Kidney Int. 2011;80:17–28.
Kremer LS, Bader DM, Mertes C, Kopajtich R, Pichler G, Iuso A, et al. Genetic diagnosis of Mendelian disorders via RNA sequencing. Nat Commun. 2017;8:15824.
Wagner M, Berutti R, Lorenz-Depiereux B, Graf E, Eckstein G, Mayr JA, et al. Mitochondrial DNA mutation analysis from exome sequencing—a more holistic approach in diagnostics of suspected mitochondrial disease. J Inherit Metab Dis. 2019;42:909–17.
Plagnol V, Curtis J, Epstein M, Mok KY, Stebbings E, Grigoriadou S, et al. A robust model for read count data in exome sequencing experiments and implications for copy number variant calling. Bioinformatics. 2012;28:2747–54.
Tsukaguchi H, Sudhakar A, Le TC, Nguyen T, Yao J, Schwimmer JA, et al. NPHS2 mutations in late-onset focal segmental glomerulosclerosis: R229Q is a common disease-associated allele. J Clin Investig. 2002;110:1659–66.
Tory K, Menyhard DK, Woerner S, Nevo F, Gribouval O, Kerti A, et al. Mutation-dependent recessive inheritance of NPHS2-associated steroid-resistant nephrotic syndrome. Nat Genet. 2014;46:299–304.
Ellard S, Baple E, Berry I, Forrester N, Turnbull C, Owens M, et al. ACGS best practice guidelines for variant classification. Association for Clinical Genomics Science, 2019.2019. https://www.acgs.uk.com/news/acgs-best-practiceguidelines-for-variant-classification-2019/.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24.
Kirby A, Gnirke A, Jaffe DB, Baresova V, Pochet N, Blumenstiel B, et al. Mutations causing medullary cystic kidney disease type 1 lie in a large VNTR in MUC1 missed by massively parallel sequencing. Nat Genet. 2013;45:299–303.
Bleyer AJ, Kmoch S, Antignac C, Robins V, Kidd K, Kelsoe JR, et al. Variable clinical presentation of an MUC1 mutation causing medullary cystic kidney disease type 1. Clin J Am Soc Nephrol. 2014;9:527–35.
Voskarides K, Damianou L, Neocleous V, Zouvani I, Christodoulidou S, Hadjiconstantinou V, et al. COL4A3/COL4A4 mutations producing focal segmental glomerulosclerosis and renal failure in thin basement membrane nephropathy. J Am Soc Nephrol. 2007;18:3004–16.
De Vriese AS, Sethi S, Nath KA, Glassock RJ, Fervenza FC. Differentiating primary, genetic, and secondary FSGS in adults: a clinicopathologic approach. J Am Soc Nephrol. 2018;29:759–74.
D’Agati VD, Fogo AB, Bruijn JA, Jennette JC. Pathologic classification of focal segmental glomerulosclerosis: a working proposal. Am J Kidney Dis. 2004;43:368–82.
Chun J, Wang M, Wilkins MS, Knob AU, Benjamin A, Bu L, et al. Autosomal dominant tubulointerstitial kidney disease-uromodulin misclassified as focal segmental glomerulosclerosis or hereditary glomerular disease. Kidney Int Rep. 2020;5:519–29.
Trautmann A, Bodria M, Ozaltin F, Gheisari A, Melk A, Azocar M, et al. Spectrum of steroid-resistant and congenital nephrotic syndrome in children: the PodoNet registry cohort. Clin J Am Soc Nephrol. 2015;10:592–600.
Giglio S, Provenzano A, Mazzinghi B, Becherucci F, Giunti L, Sansavini G, et al. Heterogeneous genetic alterations in sporadic nephrotic syndrome associate with resistance to immunosuppression. J Am Soc Nephrol. 2015;26:230–6.
Buscher AK, Kranz B, Buscher R, Hildebrandt F, Dworniczak B, Pennekamp P, et al. Immunosuppression and renal outcome in congenital and pediatric steroid-resistant nephrotic syndrome. Clin J Am Soc Nephrol. 2010;5:2075–84.
Savige J, Gregory M, Gross O, Kashtan C, Ding J, Flinter F. Expert guidelines for the management of Alport syndrome and thin basement membrane nephropathy. J Am Soc Nephrol. 2013;24:364–75.
Park E, Ahn YH, Kang HG, Yoo KH, Won NH, Lee KB, et al. COQ6 mutations in children with steroid-resistant focal segmental glomerulosclerosis and sensorineural hearing loss. Am J Kidney Dis. 2017;70:139–44.
Korkmaz E, Lipska-Zietkiewicz BS, Boyer O, Gribouval O, Fourrage C, Tabatabaei M, et al. ADCK4-associated glomerulopathy causes adolescence-onset FSGS. J Am Soc Nephrol. 2016;27:63–68.
Ashraf S, Gee HY, Woerner S, Xie LX, Vega-Warner V, Lovric S, et al. ADCK4 mutations promote steroid-resistant nephrotic syndrome through CoQ10 biosynthesis disruption. J Clin Investig. 2013;123:5179–89.
Montini G, Malaventura C, Salviati L. Early coenzyme Q10 supplementation in primary coenzyme Q10 deficiency. N Engl J Med. 2008;358:2849–50.
Preston R, Stuart HM, Lennon R. Genetic testing in steroid-resistant nephrotic syndrome: why, who, when and how? Pediatr Nephrol. 2019;34:195–210.
Guaragna MS, Lutaif AC, Bittencourt VB, Piveta CS, Soardi FC, Castro LC, et al. Frasier syndrome: four new cases with unusual presentations. Arquivos Brasileiros de Endocrinologia Metabologia. 2012;56:525–32.
Gast C, Pengelly RJ, Lyon M, Bunyan DJ, Seaby EG, Graham N, et al. Collagen (COL4A) mutations are the most frequent mutations underlying adult focal segmental glomerulosclerosis. Nephrol Dialysis Transplant. 2016;31:961–70.
Chatterjee R, Hoffman M, Cliften P, Seshan S, Liapis H, Jain S. Targeted exome sequencing integrated with clinicopathological information reveals novel and rare mutations in atypical, suspected and unknown cases of Alport syndrome or proteinuria. PLoS ONE. 2013;8:e76360.
Buscher AK, Celebi N, Hoyer PF, Klein HG, Weber S, Hoefele J. Mutations in INF2 may be associated with renal histology other than focal segmental glomerulosclerosis. Pediatr Nephrol. 2017;33:433–7.
Barua M, Brown EJ, Charoonratana VT, Genovese G, Sun H, Pollak MR. Mutations in the INF2 gene account for a significant proportion of familial but not sporadic focal and segmental glomerulosclerosis. Kidney Int. 2013;83:316–22.
Barbaux S, Niaudet P, Gubler MC, Grunfeld JP, Jaubert F, Kuttenn F, et al. Donor splice-site mutations in WT1 are responsible for Frasier syndrome. Nat Genet. 1997;17:467–70.
Acknowledgements
We would like to thank the patients and their families for their participation in the study. This work was supported by the German Research Foundation (DFG) and the Technical University of Munich (TUM) in the framework of the Open Access Publishing Program. Open access funding provided by Projekt DEAL.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Braunisch, M.C., Riedhammer, K.M., Herr, PM. et al. Identification of disease-causing variants by comprehensive genetic testing with exome sequencing in adults with suspicion of hereditary FSGS. Eur J Hum Genet 29, 262–270 (2021). https://doi.org/10.1038/s41431-020-00719-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-020-00719-3
This article is cited by
-
Pathogenic variant detection rate by whole exome sequencing in Thai patients with biopsy-proven focal segmental glomerulosclerosis
Scientific Reports (2023)
-
Ocular manifestations of the genetic causes of focal and segmental glomerulosclerosis
Pediatric Nephrology (2023)
-
Genetik in der Nephrologie
Der Nephrologe (2020)
