
Abstract
Duchenne muscular dystrophy (DMD) is caused by pathogenic variants in the DMD gene leading to the lack of dystrophin. Variability in the disease course suggests that other factors influence disease progression. With this study we aimed to identify genetic factors that may account for some of the variability in the clinical presentation. We compared whole-exome sequencing (WES) data in 27 DMD patients with extreme phenotypes to identify candidate variants that could affect disease progression. Validation of the candidate SNPs was performed in two independent cohorts including 301 (BIO-NMD cohort) and 109 (CINRG cohort of European ancestry) DMD patients, respectively. Variants in the Tctex1 domain containing 1 (TCTEX1D1) gene on chromosome 1 were associated with age of ambulation loss. The minor alleles of two independent variants, known to affect TCTEX1D1 coding sequence and induce skipping of its exon 4, were associated with earlier loss of ambulation. Our data show that disease progression of DMD is affected by a new locus on chromosome 1 and demonstrate the possibility to identify genetic modifiers in rare diseases by studying WES data in patients with extreme phenotypes followed by multiple layers of validation.
Similar content being viewed by others


Introduction
Duchenne muscular dystrophy (DMD) is the most frequent childhood onset muscular dystrophy with an incidence of 1 in 5000 newborn males [1]. DMD is characterised by progressive loss of muscle mass and function [2], with most patients dying prematurely between the 2nd and 4th decade of life [3]. DMD is caused by pathogenic variants in the DMD gene, most commonly out-of-frame deletions, either de novo or X-linked recessively inherited, that result in the absence of the gene product called dystrophin [4, 5]. DMD natural disease course is well-documented in literature with disease milestones such as loss of ambulation, dilated cardiomyopathy and respiratory complications, which significantly affect the survival of affected individuals [6, 7]. Recent re-evaluation of disease natural history with several quantitative tests [8,9,10,11,12,13,14] enabled the identification of patients progressing faster than others [11, 13,14,15,16]. Several studies have been carried out to identify the genetic factors responsible for the observed differences. This work revealed that both the deletion site (cis effect) as well as variants in other genes (trans effect) could influence the disease course. Specifically, it was observed that patients carrying out-of-frame deletions flanking exon 44 experience a longer ambulatory phase compared with patients carrying other out-of-frame deletions [15]. The milder phenotype of exon 44 skippable patients is attributed to the higher residual dystrophin production of these patients compared with other DMD genotypes [15, 17]. Other genes acting in trans were recently described as genetic modifiers of DMD, namely SPP1 (chromosome 4), LTBP4 (chromosome 19) and CD40 (chromosome 20) [18,19,20]. The first variant to be described is a non-coding SNP located 5 nucleotides upstream the transcription start site of the SPP1 gene encoding osteopontin [16, 18, 19, 21, 22]. LTBP4 was first identified in a murine model of muscular dystrophy and then validated in several independent DMD cohorts [16, 19, 22, 23]. CD40 was identified as a modifier locus by genotyping functionally relevant SNPs with an exome chip, and selecting candidate genes with a biological role in pro-inflammatory and pro-fibrotic pathways [20]. None of these modifiers has been identified by conducting a genome-wide association study (GWAS), due to the low prevalence of the disease, which precluded the design of a properly powered discovery study. Recently a GWAS performed in 253 patients with dystrophinopathy identified THBS1 (chromosome 15) as a potent DMD modifier, showing that genome-wide significance is achievable with relatively low numbers [24]. To overcome the power limitation we took a different approach based on deep genotyping of patients with extremely different clinical presentations, followed by validation in two large multicentre cohorts. To identify candidate genetic modifying variants, we performed whole-exome sequencing (WES) of patients affected by DMD with extreme phenotypes. We then validated the candidate variants in a multi-centre European cohort (Bio-NMD cohort) composed of 301 patients with DMD. The analysis has enabled us to identify 11 SNPs associated with age of ambulation loss. To further validate the identified SNPs, we explored the association with age of ambulation loss in a second independent American DMD cohort, which confirmed our findings. This enabled us to identify a locus on chromosome 1 influencing disease progression in patients with DMD.
Materials and methods
Study participants
Discovery cohorts
This study comprised a discovery phase, and a validation phase.
Comparisons of patients with extreme phenotypes were performed during the discovery phase, including patients characterised by:
-
early ambulation loss (before 8.5 years of age, n = 5) in contrast to patients who retained the ability to walk longer than average (loss of ambulation after 12 years of age, n = 5) (ambulation group),
-
early and severe cardiomyopathy, defined as an ejection fraction <40% or fraction shortening <15% (before 13 years of age, n = 6) in contrast to patients who were alive at the age of 28 with no detectable or only mild heart involvement, defined as an ejection fraction between 45 and 54% or fraction shortening between 20 and 27% (n = 11) (cardiomyopathy group)
Table 1 shows the characteristics of patients involved in both studies. Pathogenic variants have been submitted to the Leiden Open Variation Database (LOVD) [25] at https://databases.lovd.nl/shared/individuals/ (ID 00265240, 00265263-00265264, 00265266-00265279, 00265281-00265285, 00265287 and 00265396).
Validation cohorts
In the validation phase, two independent cohorts were studied. The first validation cohort was a multi-centre European cohort composed of 301 patients with DMD hereafter referred to as Bio-NMD cohort. The second validation cohort was composed of 109 individuals with DMD of European or European-American ancestry (selected by multidimensional scaling of genome-wide SNP genotyping data as previously described [16, 20]) participating in a DMD natural history study [8] (CINRG cohort). Table 2 shows the characteristics of the two validation cohorts.
Local Research Ethics Committees of all participating institutions (Leiden University Medical Center, University College London, Newcastle University, University of Ferrara and University of Montpellier) approved the study prior to its start. Informed consent for anonymised use of patients’ data was obtained for all patients. All methods were performed in accordance with the relevant institutional and country guidelines and regulations.
Identification of candidate genetic modifiers
Samples included in the discovery cohorts were prepared for sequencing using Agilent Sure Select AllExon 50 Mb XT kit according to the manufacturer’s protocol.
The prepared exome libraries for the ambulation group were sequenced on Illumina GAIIx with 2 × 76 bp number of cycles (pipeline version OLB 1.8, Casava 1.7). The average coverage of the sequenced regions was 50×. The analysis of the sequencing reads was performed using General Application Pipeline for Second generation Sequencing (GAPSS3) developed by the Department of Human Genetics, LUMC, The Netherlands. GAPSS3 performs quality control of the input data, alignment of the improved data using Stampy [26] and variant calling through SAMtools [27].
The exome libraries for the cardiomyopathy group were sequenced on an Illumina HiSeq with 100 bp paired-end. The average coverage of the sequenced regions was 54×. The data analysis was performed in-house with alignment of the data using Novoalign (www.novocraft.com) and variant calling through SAMtools.
The functional annotation of the genetic variants for both studies was performed using ANNOVAR software tool [28].
Variants showing concordant genotypes were filtered out. The criteria for selecting a SNP as a candidate genetic modifier were: (1) exonic non-synonymous SNPs; (2) the minor allele should be found in only one group; (3) and it should be present in at least 50% of the cases in the specific group. All filtering steps were performed using ANNOVAR software tool.
The selected candidate genetic modifiers were subject to further technical validation using AmpliSeq Ion Torrent Technology (Life Technologies). Custom panels with primer pairs covering the selected SNPs were designed and analysed in the same DNA samples were used in the WES discovery step.
Validation of candidate SNPs
The Sequenom (Sequenom Inc, San Diego, California, USA) MassARRAY platform was used to validate the genetic associations in the BIO-NMD cohort. Platform and software were used according to the manufacturer’s protocols. The protocol described by van den Bergen et al. [22] was used to assay all SNPs. Primers for multiplex genotyping assays were designed and sequences are available upon request. Genotyping of the CINRG cohort was performed using the Illumina Human Exome Chip previously reported [16, 20].
Statistical analysis
Cox regression was used to identify association of SNPs with time to ambulation loss. An additive genetic model was used throughout to determine genotype effects. Corticosteroid use, cohort (Ferrara, Leiden, London, Montpellier and Newcastle) and the interaction between genotype and corticosteroid use were included in the analysis as covariates. Cox regression was also used to validate SNP associations in the CINRG cohort, with an additive genetic model and a covariate for corticosteroid treatment (at least 1 year of treatment while ambulatory). Global P values were calculated using the tail-strength method [29], the P-min and the SKAT [30] statistics. In short, the tail-strength measures how much P values in a set differ from the expected uniform distribution under the null hypothesis and sums up these differences into a single test statistic. The tail-strength is powerful when many small effects exist in the data [29]. The P-min test usually performs well when a few stronger signals are present in the data. Since SNPs are not independent, empirical P values were computed using permutations for the tail strength and P-min statistics. SNPs were permuted as a block. This keeps the relationship between genotypes intact as well as between the other covariates and outcome but breaks the relationship between genotypes and outcome. Individual tests were based on a cox-model (coxph) and 1 × 104 permutations were used. Computations were parallelised using package parallelise.dynamic [31]. Global P values were computed using R version 3.2. The SKAT statistic does not assume independence of SNPs and was computed using bioconductor package globaltest [32] without permutations. We also computed the statistics SKAT standardised which is the SKAT statistics computed on standardised genotypes. Statistical analysis of genotype data for the top 10 SNPs in the CINRG cohort was performed using Cox regression of age at loss of ambulation with an additive SNP effect and a covariate for corticosteroid use (at least 1 year while ambulatory vs less or untreated). The interaction term between genotype and corticosteroid treatment was not included in the analysis of the CINRG cohort due to the low minor allele frequency of the ten variants selected for replication. Variants associated with loss of ambulation were submitted to the LOVD at https://databases.lovd.nl/shared/individuals/ (ID 00264110-00264115 and 00265391-00265395).
Bioinformatic analysis was performed by using the GeneTrail2 [33] and GTex [34] online tools. GeneTrail2 was used to test whether any of the known MEOX2 binders could be linked to the pathological pathways known to be altered in DMD. The list of MEOX2 binders (including experimental and predicted binding proteins) was obtained by Integrated Interaction Database [35] and used as input in GeneTrail2. Over-representation analysis using all supported human Uniprot IDs as background was performed. Only pathways obtained from Reactome were tested. A two-sided test followed by multiple testing correction by Benjamini and Hochberg false discovery rate (FDR) was performed and a FDR < 5% was considered significant. To assess whether the variants in the TCTEX1D1 gene were previously associated with the expression of neighbouring genes in muscle tissue, the GTex database was consulted. The rs number of the two variants was submitted and the significant expression quantitative trait loci (eQTLs) for muscle and heart tissues are reported.
Results
Identification of candidate genetic modifiers
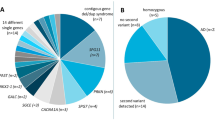
We performed WES in 27 patients with DMD with extreme phenotypes in order to identify candidate genetic modifiers. Subjects are described in Table 1. In the first comparison the SNPs genotypes in five patients characterised by early loss of ambulation (ELoA) were compared with the ones obtained in five patients with late loss of ambulation (LLoA) (Fig. 1a). About 16,000 exonic variants were analysed in each group. After filtering out the SNPs with concordant genotype between groups, intronic SNPs, synonymous SNPs and SNPs present in <3 cases per group, we were left with 104 SNPs associated with ELoA and 51 SNPs associated with LLoA. Samples resequencing by Ion Torrent confirmed the genotype of 95 ELoA SNPs and 46 LLoA SNPs.

a DMD patients with early loss of ambulation (before the age of 8.5 years) and late loss of ambulation (after 12 years). b DMD patients with late onset cardiomyopathy and patients with early onset of cardiomyopathy. WES whole-exome sequencing, LoA loss of ambulation, GT genotype, NS non-synonymous.
In the second comparison, six patients with DMD with early cardiomyopathy (ECM) were compared with eleven patients with long survival and no substantial cardiomyopathy (LS) (Fig. 1b). More than 22,000 exonic variants were analysed in each group. After filtering out SNPs following the same criteria as above, 62 SNPs were associated with ECM and 57 SNPs with LS. Resequencing of DNA samples confirmed 47 ECM SNPs and 54 LS SNPs. All SNPs that passed the technical validation were selected for further validation.
Validation of candidate SNPs
A total of 242 SNPs entered the validation phase, 199 of which were successfully genotyped in the first validation cohort consisting of 301 patients with DMD (Bio-NMD cohort, described in the methods section and in Table 2). The remaining 43 SNPs were excluded as they did not fit in the plate design or due to the sequence context which did not allow genotyping of those positions. SNPs were excluded before data analysis for minor allele frequencies below 10%, for violation of the Hardy Weinberg equilibrium or for high genotype missingness (top 5% of SNPs with highest percentage of missing data). The remaining 121 SNPs provided in Supplementary Table 1 were included in the analysis. Under P-min model, there was no single SNP strongly associated with age of ambulation loss. However, the tail strength, SKAT model and SKAT standardised models showed significant association for 11 SNPs with age of ambulation loss (P < 0.05 for all three models). A summary of the results obtained is presented in Table 3. The Q–Q plot presented in Fig. 2 shows an enrichment of observed P values compared with the expected ones, which is to be expected in the validation phase where candidate genes are analysed. Of note, SNPs in complete linkage disequilibrium (LD) were identified at two different loci. Interestingly, for six SNPs a significant interaction with corticosteroid treatment was found (Table 3).

The observed P values are lower compared with the expected ones showing an enrichment in the obtained distribution compared with the observed one.
External validation confirms an association with a locus on chromosome 1
To further validate the identified associations, we investigated the 11 SNPs confirmed in the Bio-NMD cohort in an independent cohort of 109 patients with DMD of European ancestry followed up in a multi-centre DMD natural history cohort (described in the methods section and in Table 2). Genotype data were already available for these patients since they were included in a recent exome chip association study involving patients with DMD [20]. Ten of eleven SNPs were present in the published dataset, while genotyping data were not available for SNP rs12146487 and therefore SNP rs2244621 in close proximity (419 bp) was considered instead. Analysis of the CINRG cohort showed that the two SNPs (rs1060575 and rs3816989) in LD at the TCTEX1D1 locus were associated with age at loss of ambulation in the CINRG cohort (P = 0.032) (Fig. 3). Both SNPs have an effect on the transcript originating from this locus. SNP rs1060575 is an A to T transversion responsible for an aminoacidic change from glutamate to aspartate in exon 3 (https://www.ncbi.nlm.nih.gov/SNP/snp_ref.cgi?type=rs&rs=rs1060575), while rs3816989 is G to A transition affecting the donor splice site of exon 4 and inducing the skipping of exon 4 [36]. The minor alleles were associated with a faster disease progression as shown by the Kaplan–Meier curves (Fig. 3).

a The effect of rs1060575 in the BIO-NMD validation cohort. b The SNP effects in the CINRG validation cohort. Censored patients are indicated by a cross on the lines.
Discussion
Patients affected by DMD experience a rather predictable disease course with known disease milestones. International collaborations and deep phenotyping have enabled to collect and analyse a large body of data that allowed the identification of different disease trajectories across patients [37, 38]. The identification of factors that affect disease progression is of interest as these factors may represent both prognostic biomarkers and potential therapeutic targets. Differences in disease progression have been connected to the deletion site within the DMD gene [15, 17] as well as to other factors such as disease modifying SNPs in SPP1, LTBP4, CD40 and THBS1 genes [18,19,20, 22, 24]. The rare disease nature of the disease has not allowed to properly power a GWA study in DMD. However the strong collaboration of international networks, as well as deep phenotyping of patients, has been the inspiration to perform an exome wide discovery study followed by multiple layers of validation. Indeed, we present here WES data of patients with DMD with extreme phenotypes based on the hypothesis that analysing homogeneous groups of patients with extreme phenotype would increase the chance to detect genetic modifiers in a small number of patients. A similar approach has been successfully used in patients affected by cystic fibrosis but never before in neuromuscular patients [39]. The discovery phase focused on two major variables responsible for DMD morbidity and mortality: skeletal muscle weakness (and loss of ambulation as its direct surrogate) and severity of cardiomyopathy. The cut-off values were chosen based on the personal experience of the paediatric neurologists participating to the BIO-NMD consortium, local clinical databases and natural history reports describing different DMD trajectories. Our recently concluded natural history studies further allowed us to refine the proposed categorisation based on estimates derived from the distribution of the events over the age in natural history studies [38]. We reached a consensus that losing ambulation before 8.5 years of age was a clinically meaningful change compared with after 12. Using this categorisation we were able to segregate patients belonging to different categories. Published estimates for loss of ambulation consistently report a mean age of 10 [22, 24, 37]. The most recent report showed a mean of 10.6 with standard deviation [SD] = 2.3 [24]. The 8.5 threshold is therefore about 1 SD below the mean. Comparable considerations have been used for the cardiomyopathy group. Comparison of extreme cases enabled to identify 242 candidate DMD modifying variants to be investigated further. A first layer of validation in a cohort of 301 patients with DMD with the whole spectrum of clinical severity, representing the largest European DMD cohort described so far, confirmed a significant association of 11 SNPs with the age of ambulation loss. Subsequent validation in the CINRG Duchenne Natural History study considering participants of European ancestry narrowed down the number of candidates to two SNPs (rs1060575 and rs3816989) in complete LD in the TCTEX1D1 locus on chromosome 1. SNP rs1060575 is a missense SNP leading to a likely benign substitution (according to SIFT [40] and PolyPhen [41]) of glutamic acid with aspartic acid in exon 3, which is also observed in other species. The second SNP rs3816989 is located at position +1 of the donor splice site of exon 4 and it has been reported to lead to TCTEX1D1 exon 4 skipping [36] reducing the resulting polypeptide chain from 179 to 72 amino acid residues. As yet the role of TCTEX1D1 is largely unknown, so it is uncertain how the lack of amino acids affects protein function. The gene is more highly expressed in brain according to the Expression Atlas (EMBL-EBI) [42], and it is expressed at lower level in skeletal muscle and heart. There is evidence for TCTEX1D1 protein interaction with MEOX2 (Mesenchyme Homeo Box 2) protein [43]. MEOX2 is expressed in skeletal muscle, it regulates muscle progenitor cells such as Pax3/7 positive cells and it is involved in limb myogenesis in vertebrates. MEOX2 null mice show hypertrophic, centrally nucleated fibres (normally found in mdx mice as a consequence of muscle regeneration) as well as a shift towards oxidative type I fibres and reduced myonuclear domains. It is over-expressed in muscle biopsies of patients with DMD as well as in patients affected by Emery–Dreifuss muscular dystrophy and juvenile dermatomyositis [44]. A pathway analysis performed with GeneTrail2 [33] including all known and potential MEOX2 binding partners (obtained from the Integrated Interaction Database [35]) revealed that MEOX2 interacting partners are involved in a number of pathways previously related to DMD pathophysiology such as NF-κB and SMAD signalling.
Both rs1060575 and rs3816989 variants were identified in the comparison between patients showing early signs of cardiomyopathy with long survivor patients, without significant cardiac involvement. Given that the discovery cohort was originally focused also on the cardiac phenotype; the association with loss of ambulation suggests that the identified gene and variants can have a role on both cardiac and skeletal muscles. To understand whether SNPs rs1060575 and rs3816989 drive the expression of the TCTEX1D1 gene and other neighbouring genes, we searched for eQTLs in the GTEx database. Interestingly, the minor alleles of these variants are associated with the reduced expression of TCTEX1D1 and an increased expression of the neighbouring gene SGIP1 in both muscle and heart (Supplementary Fig. 1). Given that the minor allele was associated with a worse outcome in both discovery and validation cohorts, one could postulate that an increased expression of SGIP1 and a decreased expression of TCTEX1D1 could be deleterious for the cardiac phenotype in patients with DMD. Interestingly, a recent GWA meta-analysis for quantitative electrocardiography traits, showed that variants in the TCTEX1D1/SGIP1 locus are associated with QT interval [45]. More research (e.g. conditional knock down of TCTEX1D1 in skeletal and cardiac muscles in animal models or study the different TCTEX1D1 proteoforms in patients’ derived cells) will be needed to provide solid mechanistic explanation of the role of these variants in DMD disease progression.
The comparison between the ELOA and LLOA did not lead to the identification of other variants. One possible cause for this is the selection of patients involved in the LLOA discovery cohort in which two patients carried out-of-frame deletions that can be reframed by exon 44 skipping. While it is now known that patients with this genomic characteristic experience a somewhat milder disease progression [15, 17], this was not known when the study was designed. An improved patients selection could have made the comparison more informative for the identification of other variants possibly acting in trans.
Our study is the first to identify modifiers associated with cardiomyopathy severity in DMD; the obtained data will now support the interpretation of studies on cardiac protection.
To the best of our knowledge, our work is the first in which exome sequencing was used to identify genetic modifiers in patients affected by DMD. In view of the relative rarity of DMD and the strategy to concentrate on the extreme phenotypes, the study was underpowered. However, multiple layers of subsequent validation enabled us to exclude false positive signals and identify TCTEX1D1 as modifier of DMD disease progression. The success of this approach could be beneficial also for other rare diseases where the small cohorts size does not enable a classical GWAS design.
References
Norwood FLM, Harling C, Chinnery PF, Eagle M, Bushby K, Straub V. Prevalence of genetic muscle disease in Northern England: in-depth analysis of a muscle clinic population. Brain. 2009;132:3175–86.
Straub V, Balabanov P, Bushby K, Ensini M, Goemans N, De Luca A, et al. Stakeholder cooperation to overcome challenges in orphan medicine development: the example of Duchenne muscular dystrophy. Lancet Neurol. 2016;15:882–90.
Bushby K, Finkel R, Birnkrant DJ, Case LE, Clemens PR, Cripe L, et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and pharmacological and psychosocial management. Lancet Neurol. 2010;9:77–93.
Ellis JA, Vroom E, Muntoni F. 195th ENMC International Workshop: newborn screening for Duchenne muscular dystrophy 14–16th December, 2012, Naarden, The Netherlands. Neuromuscul Disord. 2013;23:682–9.
Aartsma-Rus A, Ginjaar IB, Bushby K. The importance of genetic diagnosis for Duchenne muscular dystrophy. J Med Genet. 2016;53:145–51.
Desguerre I, Christov C, Mayer M, Zeller R, Becane H-M, Bastuji-Garin S, et al. Clinical heterogeneity of duchenne muscular dystrophy (DMD): definition of sub-phenotypes and predictive criteria by long-term follow-up. PLoS ONE. 2009;4:e4347.
Magri F, Govoni A, D’Angelo MG, Del Bo R, Ghezzi S, Sandra G, et al. Genotype and phenotype characterization in a large dystrophinopathic cohort with extended follow-up. J Neurol. 2011;258:1610–23.
Henricson EK, Abresch RT, Cnaan A, Hu F, Duong T, Arrieta A, et al. The cooperative international neuromuscular research group Duchenne natural history study: glucocorticoid treatment preserves clinically meaningful functional milestones and reduces rate of disease progression as measured by manual muscle testing and other. Muscle Nerve. 2013;48:55–67.
Henricson E, Abresch R, Han JJ, Nicorici A, Goude Keller E, de Bie E et al. The 6-minute walk test and person-reported outcomes in boys with duchenne muscular dystrophy and typically developing controls: longitudinal comparisons and clinically-meaningful changes over one year. PLoS Curr. 2013. https://doi.org/10.1371/currents.md.9e17658b007eb79fcd6f723089f79e06.
Mcdonald CM, Henricson EK, Abresch RT, Florence JM, Eagle M, Gappmaier E, et al. THE 6-minute walk test and other endpoints in Duchenne muscular dystrophy: Longitudinal natural history observations over 48 weeks from a multicenter study. Muscle Nerve. 2013;48:343–56.
McDonald CM, Henricson EK, Abresch RT, Florence J, Eagle M, Gappmaier E, et al. The 6-minute walk test and other clinical endpoints in duchenne muscular dystrophy: reliability, concurrent validity, and minimal clinically important differences from a multicenter study. Muscle Nerve. 2013;48:357–68.
Mazzone ES, Messina S, Vasco G, Main M, Eagle M, D’Amico A, et al. Reliability of the north star ambulatory assessment in a multicentric setting. Neuromuscul Disord. 2009;19:458–61.
Mazzone E, Vasco G, Sormani MP, Torrente Y, Berardinelli A, Messina S, et al. Functional changes in Duchenne muscular dystrophy: a 12-month longitudinal cohort study. Neurology. 2011;77:250–6.
Ricotti V, Ridout DA, Pane M, Main M, Mayhew A, Mercuri E et al. The NorthStar Ambulatory Assessment in Duchenne muscular dystrophy: considerations for the design of clinical trials. J Neurol Neurosurg Psychiatry. 2015. https://doi.org/10.1136/jnnp-2014-309405.
van den Bergen JC, Ginjaar HB, Niks EH, Aartsma-Rus AVJJGM, van den Bergen J, Ginjaar H. Prolonged ambulation in Duchenne patients with a mutation amenable to exon 44 skipping. J Neuromuscul Dis. 2014;1:91–94.
Bello L, Kesari A, Gordish-Dressman H, Cnaan A, Morgenroth LP, Punetha J, et al. Genetic modifiers of ambulation in the cooperative international neuromuscular research group Duchenne natural history study. Ann Neurol. 2015;77:684–96.
Anthony K, Arechavala-Gomeza V, Ricotti V, Torelli S, Feng L, Janghra N, et al. Biochemical characterization of patients with in-frame or out-of-frame DMD deletions pertinent to exon 44 or 45 skipping. JAMA Neurol. 2014;71:32.
Pegoraro E, Hoffman EP, Piva L, Gavassini BF, Cagnin S, Ermani M, et al. SPP1 genotype is a determinant of disease severity in Duchenne muscular dystrophy. Neurology. 2011;76:219–26.
Flanigan KM, Ceco E, Lamar KM, Kaminoh Y, Dunn DM, Mendell JR, et al. LTBP4 genotype predicts age of ambulatory loss in duchenne muscular dystrophy. Ann Neurol. 2013;73:481–8.
Bello L, Flanigan KM, Weiss RB, United Dystrophinopathy Project, Spitali P, Aartsma-Rus A, et al. Association study of exon variants in the NF-κB and TGFβ pathways identifies CD40 as a modifier of duchenne muscular dystrophy. Am J Hum Genet. 2016;99:1163–71.
Bello L, Piva L, Barp A, Taglia A, Picillo E, Vasco G, et al. Importance of SPP1 genotype as a covariate in clinical trials in Duchenne muscular dystrophy. Neurology. 2012;79:159–62.
van den Bergen JC, Hiller M, Böhringer S, Vijfhuizen L, Ginjaar HB, Chaouch A et al. Validation of genetic modifiers for Duchenne muscular dystrophy: a multicentre study assessing SPP1 and LTBP4 variants. J Neurol Neurosurg Psychiatry. 2015;86:1060–5.
Lamar K, McNally EM. Genetic modifiers for neuromuscular diseases. J Neuromuscul Dis. 2014;1:3–13.
Weiss RB, Vieland VJ, Dunn DM, Kaminoh Y, Flanigan KM. Long-range genomic regulators of THBS1 and LTBP4 modify disease severity in duchenne muscular dystrophy. Ann Neurol. 2018;84:234–45.
Fokkema IFaC, Taschner PEM, Schaafsma GCP, Celli J, Laros JFJ, den Dunnen JT. LOVD v.2.0: the next generation in gene variant databases. Hum Mutat. 2011;32:557–63.
Lunter G, Goodson M. Stampy: a statistical algorithm for sensitive and fast mapping of Illumina sequence reads. Genome Res. 2011;21:936–9.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The sequence alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–9.
Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164.
Taylor J. A tail strength measure for assessing the overall univariate significance in a dataset. Biostatistics. 2005;7:167–81.
Wu MC, Lee S, Cai T, Li Y, Boehnke M, Lin X. Rare-variant association testing for sequencing data with the sequence kernel association test. Am J Hum Genet. 2011;89:82–93.
Böhringer S. Dynamic parallelization of R functions. R J. 2013;5:88–96.
Goeman JJ, van de Geer SA, van Houwelingen HC. Testing against a high dimensional alternative. J R Stat Soc Ser B. 2006;68:477–93.
Stöckel D, Kehl T, Trampert P, Schneider L, Backes C, Ludwig N, et al. Multi-omics enrichment analysis using the GeneTrail2 web service. Bioinformatics. 2016;32:1502–8.
Consortium* TGte. The genotype-tissue expression (GTEx) project. Nat Genet. 2013;45:580.
Kotlyar M, Pastrello C, Sheahan N, Jurisica I. Integrated interactions database: tissue-specific view of the human and model organism interactomes. Nucleic Acids Res. 2016;44:D536–41.
ElSharawy A, Manaster C, Teuber M, Rosenstiel P, Kwiatkowski R, Huse K, et al. SNPSplicer: systematic analysis of SNP-dependent splicing in genotyped cDNAs. Hum Mutat. 2006;27:1129–34.
Koeks Z, Bladen CL, Salgado as D, van Zwet E, Pogoryelova O, McMacken G, et al. Clinical outcomes in duchenne muscular dystrophy: a study of 5345 patients from the TREAT-NMD DMD global database. J Neuromuscul Dis. 2017;4:293–306.
Muntoni F, Domingos J, Manzur AY, Mayhew A, Guglieri M, Sajeev G, et al. Categorising trajectories and individual item changes of the North Star Ambulatory Assessment in patients with Duchenne muscular dystrophy. PLoS ONE. 2019;14:e0221097.
Emond MJ, Louie T, Emerson J, Zhao W, Mathias RA, Knowles MR, et al. Exome sequencing of extreme phenotypes identifies DCTN4 as a modifier of chronic Pseudomonas aeruginosa infection in cystic fibrosis. Nat Genet. 2012;44:886–9.
Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4:1073–82.
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–9.
Petryszak R, Burdett T, Fiorelli B, Fonseca NA, Gonzalez-Porta M, Hastings E et al. Expression Atlas update—A database of gene and transcript expression from microarray—and sequencing-based functional genomics experiments. Nucleic Acids Res. 2014; 42. https://doi.org/10.1093/nar/gkt1270.
Rolland T, Taşan M, Charloteaux B, Pevzner SJ, Zhong Q, Sahni N, et al. A proteome-scale map of the human interactome network. Cell. 2014;159:1212–26.
Bakay M. Nuclear envelope dystrophies show a transcriptional fingerprint suggesting disruption of Rb-MyoD pathways in muscle regeneration. Brain. 2006;129:996–1013.
van Setten J, Verweij N, Mbarek H, Niemeijer MN, Trompet S, Arking DE et al. Genome-wide association meta-analysis of 30,000 samples identifies seven novel loci for quantitative ECG traits. Eur J Hum Genet. 2019: 952–62.
Acknowledgements
We acknowledge Raimo Tanzi and Vincent Pignol for the technical support for sequencing and data analysis. We acknowledge Dr Teresinha Evangelista, Dr Michela Guglieri, Dr Chiara Marini-Bettolo, Dr Antonio Atalaia, Dr Volker Straub and Dr Kate Bushby for following up the patients and collecting samples. We acknowledge Dr Stanley F. Nelson and Dr Richard T. Wang (UCLA), and Dr Akanchha Kesari and Dr Jaya Punetha for generating genotype data for the CINRG cohort. We acknowledge all CINRG investigators for collecting the clinical data and samples used for the validation. This work was supported by the European Commission FP7 funded grant “Bio-NMD: Identifying and validating pre-clinical biomarkers for diagnostics and therapeutics of Neuromuscular Disorders” (HEALTH-2009-2.4.4-1: Rare neurological diseases), grant agreement 241665. The work was also funded by the European Community’s Seventh Framework Programme (FP7/2007–2013) under grant agreement n° 305121 “Integrated European Project on Omics Research of Rare Neuromuscular and Neurodegenerative Diseases (NEUROMICS)” and grant agreement n° 305444 “RD-Connect”. PS would like to acknowledge the Association Française contre les Myopathies (grants n. 17013 and 17724) for supporting this work. FM is supported by the National Institute for Health Research Biomedical Research Centre at Great Ormond Street Hospital for Children NHS Foundation Trust and University College London. The support of the Muscular Dystrophy UK to the Dubowitz Neuromuscular Centre and of the MRC Neuromuscular Centre Biobank is also gratefully acknowledged. This study was also partially supported by the NIHR grant “Rare Diseases Translational Research Collaboration” on Duchenne muscular dystrophy to FM. CINRG was supported by grants from the US Department of Defense (grant #W81XWH-12-1-0417), US Department of Education/NIDRR (grants #H133B031118, #H133B090001), and US National Institutes of Health/NIAMS (grant #R01AR061875). SC is supported by the German Research Foundation (DFG CI 218/1–1). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
CINRG Investigators
Alberto Dubrovsky, Andrew Kornberg, Kathryn North, Monique Ryan, Richard Webster, W. Douglas Biggar, Laura C. McAdam, Jean K. Mah, Hanna Kolski, V. Vishwanathan, S. Chidambaranathan, Yoram Nevo, Ksenija Gorni, Jose Carlo, Mar Tulinius, Timothy Lotze, Tulio E. Bertorini, John W. Day, Peter Karachunski, Paula R. Clemens, Hoda Abdel-Hamid, Jean Teasley, Nancy Kuntz, Sherilyn Driscoll, John B. Bodensteiner, Anne M. Connolly, Alan Pestronk, R. T. Abresch, Erik K. Henricson, Nanette C. Joyce, Craig M. McDonald, Avital Cnaan, Heather Gordish-Dressmsn, Lauren P. Morgenroth, Robert Leshner, Carolina Tesi-Rocha, Mathula Thangarajh, Tina Duong
Author information
Authors and Affiliations
Consortia
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Spitali, P., Zaharieva, I., Bohringer, S. et al. TCTEX1D1 is a genetic modifier of disease progression in Duchenne muscular dystrophy. Eur J Hum Genet 28, 815–825 (2020). https://doi.org/10.1038/s41431-019-0563-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-019-0563-6
This article is cited by
-
A genome-wide association analysis of loss of ambulation in dystrophinopathy patients suggests multiple candidate modifiers of disease severity
European Journal of Human Genetics (2023)
-
Is it time for genetic modifiers to predict prognosis in Duchenne muscular dystrophy?
Nature Reviews Neurology (2023)
-
Duchenne muscular dystrophy
Nature Reviews Disease Primers (2021)
-
Evaluating the potential of novel genetic approaches for the treatment of Duchenne muscular dystrophy
European Journal of Human Genetics (2021)
-
Transcriptome-wide association study identifies new susceptibility genes and pathways for depression
Translational Psychiatry (2021)
