
Abstract
Congenital hearing impairment (HI) is the most common sensory impairment and can be isolated or part of a syndrome. Diagnosis through newborn hearing screening and management through early intervention, hearing aids and cochlear implantation is well established in the Australian setting; however understanding the genetic basis of congenital HI has been missing. This population-derived cohort comprised infants with moderate-profound bilateral HI born in the 2016–2017 calendar years, detected through newborn hearing screening. Participants were recruited through an integrated paediatric, otolaryngology and genetics HI clinic and offered whole exome sequencing (WES) on a HiSeq4000 or NextSeq500 (Illumina) platform with a targeted average sequencing depth of 100x and chromosome microarray on the Illumina Infinium core exome-24v1.2 platform. Of those approached, 68% (106/156) consented to participate. The rate of genetic diagnosis was 56% (59/106), significantly higher than standard of care (GJB2/6 sequencing only), 21% (22/106). There were clinical implications for the 106 participants: 36% required no further screening, 9% had tailored screening initiated, 2% were offered treatment and 4% had informed care for a complex neurodevelopmental syndrome. WES in this cohort demonstrates the range of diagnoses associated with congenital HI and confirms the genetic heterogeneity of congenital HI. The high diagnostic yield and clinical implications emphasises the need for genomic sequencing to become standard of care.
Similar content being viewed by others

Introduction
The diagnosis and management of infants with congenital hearing impairment (HI) has evolved with the introduction of newborn hearing screening. However, in many Australian children with HI, a specific genetic cause is not identified, due to the genetic heterogeneity of HI and lack of access to funded genomic testing. Meanwhile, genomic sequencing has allowed the characterisation of the molecular causes of HI in several large clinic ascertained cohorts [1,2,3] and many smaller cohorts [4,5,6,7,8,9,10,11,12], resulting in the recommendation of genomic sequencing as the gold standard for genetic diagnosis of HI [13].
The rate of genetic diagnoses in previous studies has varied widely, from 10 to 80% (summarised in [14]), according to the participant ascertainment and testing methodology. A higher diagnostic yield from genomic testing has been associated with inclusion of patients with early onset of hearing loss [2, 9, 10], patients who have not had previous genetic testing for common genes such as GJB2 [4,5,6,7, 9, 12], and patients with suspected genetic syndromes [1, 4]. As expected, diagnostic yield also increases with the inclusion of greater numbers of deafness genes [14, 15] and the addition of analysis for copy number variation (CNV) [16,17,18].
We aimed to characterise the molecular causes of congenital HI in a population-derived 2-year birth cohort of infants, diagnosed with congenital HI by newborn hearing screening. We sought to investigate the diagnostic utility of whole exome sequencing (WES) and chromosome microarray as a first line testing regime and evaluate the effect of early diagnosis on the clinical management of this cohort.
Materials and methods
The methods are described in detail in the protocol publication [19] and are summarised below.
Study design and participants
Infants born in the state of Victoria, Australia in 2016 or 2017 calendar years and diagnosed with bilateral moderate, severe or profound [20] HI at any point in time within the 2-year study period were eligible to participate. Screening through the Victorian Infant Hearing Screening Program was a pre-requisite for participation. Infants classified as having unilateral, mild or conductive HI were excluded. For the majority of participants (both metropolitan and regional) recruitment was coordinated through an integrated paediatric, otolaryngology and genetics HI clinic. The remainder were recruited in regional Victoria via a clinical genetics outreach service. All participants received genetic counselling. Any participant with additional medical problems, dysmorphic features or a complex family history was also assessed by a clinical geneticist.
The paediatric clinics provided investigation and management as per their usual standard of care, which typically included sequencing of GJB2/6, magnetic resonance imaging (MRI) to assess inner ear anatomy, audiology testing for first degree family members, ophthalmology assessment, testing for congenital cytomegalovirus (CMV), and electrocardiogram (ECG) for infants with bilateral profound HI [21]. In the study cohort, analysis of GJB2/6 was performed through WES and microarray rather than individual gene sequencing which was the standard of care prior to this study.
Genetic testing
WES was performed in a clinically accredited laboratory on either a HiSeq4000 or NextSeq500 (Illumina) with a targeted average sequencing depth of 100×. For all participants, a targeted deafness panel was used for analysis, comprising 144 genes, and including genes known to cause non-syndromic and syndromic HI. In participants suspected of having a syndrome diagnosis that was not included in the 144 deafness genes, a phenotype-driven list of genes was included in the analysis. Variants were classified according to the principles outlined in the American College of Medical Genetics and Genomics (ACMG) and Association for Molecular Pathology standards for interpretation of sequence variants [22]. Variant classification was reviewed in a multidisciplinary team meeting attended by clinical geneticists, medical subspecialists, genetic counsellors, medical scientists and bioinformaticians. Variants of uncertain significance (VUS) were sub-classified according to the available evidence toward pathogenicity or otherwise [23]. VUS ‘favour pathogenic’ (VUS-FP) variants were accounted for in the analysis as having achieved a genetic diagnosis if they were in trans with another VUS-FP, likely pathogenic (LP) or pathogenic (P) variant and there was no conflicting benign evidence. If consent and samples could be obtained, variants were tested in parents to determine if they were de novo or inherited, in cis or in trans. The mitochondrial genome was not available to be examined in this cohort. All participants had a microarray performed to detect CNVs. Microarray analysis was performed using the Illumina Infinium core exome-24v1.2 platform that has a total of 550 K of markers/SNPs/probes at an average interval of 5–10 kb through the genome. All variants detected were submitted to ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) for sequence variants and Decipher for CNV’s (https://decipher.sanger.ac.uk/).
Results
Recruitment
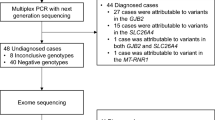
One hundred and sixty-nine patients were eligible to participate and, of the 156 patients who were contacted, 106 (68%) consented to WES and microarray. Thirteen patients were not contacted: five were unable to be reached, one had already had WES, one was deceased and six did not have a diagnosis of HI consistent with the inclusion criteria on review of their audiology testing. Participants were recruited between 4 weeks and 1 year of age and all received results prior to 2 years of age. A flowchart demonstrates recruitment of patients (Fig. 1).

A flowchart demonstrating recruitment of patients.
Of the 50 parents who declined testing for their child, 20 (40%) declined at the initial telephone call, 15 (30%) after receiving written information after the call and the remaining 15 (30%) following a face-to-face appointment [19]. Reasons for declining are summarised in Table 1. Eight of those who declined gave the reason that they had received a genetic diagnosis for their child prior to being contacted about the study: these included GJB2 variants, STRC deletion, X chromosome deletion involving the CASK gene, peroxisomal biogenesis disorders PEX26, PEX7 and Pallister–Killian syndrome. Five cited they had another explanation for their child’s HI: these included congenital CMV and prematurity. Six were concerned about insurance, privacy, research or having a blood sample collected and nine stated they felt too ‘overwhelmed’ to participate. The remainder did not provide a reason for declining participation. A firsthand experience with HI was disclosed in 12% (6/50). Three of these were hearing-impaired parents, two had another child with HI and one parent had two siblings with HI but reported normal hearing. The degree of HI in the cohort that declined testing was 56% moderate HI, 36% severe HI and 8% profound HI.
Participant demographics and characteristics
Details of participants are summarised in Table 2. There was a slight excess of male infants. Moderate HI was diagnosed in 54% of cases making it more common than severe HI and profound HI. There was a family history of congenital or early childhood HI in a parent or sibling in 15% of cases. The majority of families identified as Australian or New Zealander, followed by European and Asian, the proportions being consistent with the ethnicities of the general Victorian population [24]. Assisted reproductive technology was used in 13% of pregnancies.
The majority of participants (74%) had no clinical symptoms or signs other than HI, while the remainder had one or more additional features. There was a higher representation, compared with the Victorian population, of participants from higher socio-economic backgrounds despite this being a population-based sample.
Diagnostic yield
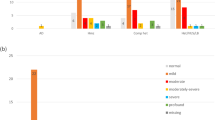
WES and microarray together resulted in a diagnostic yield of 56% (59/106). Amongst these, 21% (22/106) had causative GJB2/GJB6 variants, 15% (16/106) had another non-syndromic HI gene and 20% (21/106) had a syndrome identified, see Fig. 2. Microarray contributed to the diagnosis in seven cases. In two cases there were a deletion and a sequence variant found in trans (GJB2/6 and OTOA) and in a further two cases microarray alone made the diagnosis (ELN and TGFBR2). In three cases a homozygous deletion involving the STRC gene was detected on microarray and confirmed using multiplex ligation-dependant probe amplification (MLPA). Amongst the syndromic diagnoses (ELN, TGFBR2, SPTB, SON), HI is not a consistent feature of these conditions. If these cases are excluded from the analysis the diagnostic yield for HI specifically is 52% (55/106). A full list of genes and variants identified is in Table 3. Five participants had congenital CMV infection confirmed from PCR of newborn bloodspot cards. One of the participants with congenital CMV infection also had compound heterozygous GJB2 pathogenic variants detected. In addition to GJB2 (21%), the most common causative genes identified were MYO15A (5%), SLC26A4 (5%) and STRC (4%). The majority of diagnoses, 81% (48/59), were autosomal recessive (AR) inheritance. Eleven cases were autosomal dominant (AD) of which eight were de novo and one inherited from a parent who also had HI. Two cases were consistent with a dominant family history but the affected parent was not available for testing. No cases of X-linked HI were identified. Six participants were classified as having auditory neuropathy; of these, five were born prematurely and required phototherapy for hyperbilirubinemia and none had a genetic diagnosis made. The rate of diagnosis was higher in severe (70%) and profound (68%) HI compared with moderate (46%).

Diagnostic yield.
Of the 16 participants who had a first degree relative with congenital HI, six received a diagnosis. These were three cases of GJB2/6 variants, one of which was consistent with AD Bart–Pumphrey syndrome, one case of Stickler syndrome that was also diagnosed clinically in a parent, two aunts and the paternal grandmother, and one of Usher syndrome type II which was confirmed subsequently in a sibling.
Three cases that were designated as having received a genetic diagnosis had a VUS-FP variant in trans with an LP or P variant, and another five cases were homozygous for a VUS-FP. If these variants were removed from the analysis the overall diagnostic rate decreased to 48% (51/106).
Of the participants that did not receive a molecular diagnosis, 26% (12/47) had a single heterozygous variant in a recessive HI gene. Six of these were classified as VUS-LP, five as LP and one as P. In these cases the coverage of the gene was manually checked to investigate the possibility of their being a second variant that was undetected.
Clinical implications
Details of the clinical implications of genetic findings are presented in Table 4. In summary: 36% (38/106) of participants received a non-syndromic HI diagnosis and were discharged from further screening or surveillance, 9% (10/106) were moved to a screening protocol tailored to their genetic diagnosis, 2% (2/106) had a specific treatment offered and 4% (4/106) had a complex neurodevelopmental syndrome diagnosed that informed medical care.
The turn-around time from parents providing consent to receiving results was highly variable from 6 weeks to 6 months, with shorter time frames achieved as the study progressed.
Recurrence risk was determined for all families whose child received a diagnosis. Eight percent (8/106) of cases had confirmed de novo variants implying low risk of recurrence, and 48% (51/106) had inherited variants and parents were informed of their reproductive options.
Six relatives of tested infants received a diagnosis through segregation analysis. This included three clinically affected parents (Stickler syndrome, Noonan syndrome and Bart–Pumphrey syndrome), one affected sibling (Usher syndrome, type II) and the parents of a participant with Leigh syndrome who, as heterozygotes, carry an increased risk of paraganglioma and pheochromocytoma [25].
Secondary findings
KCNE1 was included in the gene list due to its association with AR Jervell and Lange-Nielson syndrome. Heterozygous variants in KCNE1 are associated with AD Long QT syndrome [26]. One participant had a single variant detected, c.226G>A, which has previously been described as pathogenic [27], and which segregated with the infant’s mother. While this does not explain the infant’s HI, he has received a full cardiac evaluation with normal QT interval on ECG and Holter monitor. His mother has also had a full cardiac evaluation and has been found to have a prolonged QT interval. The infant is receiving cardiology follow-up and consideration of beta blocker therapy, and his siblings are being assessed.
Discussion
WES in this cohort demonstrates the wide range of diagnoses associated with congenital HI and confirms the genetic heterogeneity of this condition. The high diagnostic yield and subsequent clinical implications for individuals and families emphasise the need for genomic sequencing to become standard of care. WES significantly increased the rate of diagnosis when compared with previous standard of care in our state (GJB2/6 sequencing), which would have identified an aetiology in only 21% (22/106) of participants. A genetic diagnosis was made in a further 35% (37/106) of participants, 16 of whom had other non-syndromic HI confirmed and 21 had a syndrome. The degree of HI in the recruited versus the declined cohort was similar, eliminating this as a source of bias in the results.
Overall 92% (54/59) of participants who received a diagnosis had some change in their clinical management, corresponding to 51% (54/106) of participants in the overall cohort. More than one third (37/106) were discharged from further screening and surveillance, thus reducing the burden on hospital resources and alleviating the ‘watch and wait’ anxiety for parents who are aware of the possibility of an evolving syndrome. Those without a molecular diagnosis continued to have surveillance and investigation as per recently published local guidelines [21]. When a syndrome was identified, management was streamlined and informed. For example, in the participant with Cockayne syndrome caused by homozygous variants in ERCC6, this diagnosis provided important information for clinical care, such as avoiding metronidazole [28] and exposure to sunlight, and allowed the family to plan for the future with the understanding that Cockayne syndrome is a life-limiting condition [29]. For the three participants with hidden syndromes that present initially as an isolated HI phenotype (GATA3, USH2A, MYO7A) early molecular diagnosis made a significant impact on surveillance, education and access to support groups.
In this cohort, eight families were reassured of a low recurrence risk due to their child’s variant being de novo and the remaining 51 families who received a diagnosis were made aware of the likelihood of recurrence and the reproductive options available to them. Variations in cultural and personal values result in vastly differing responses to the birth of an infant with HI. However, access to prenatal diagnosis or pre-implantation genetic testing is sought by a number of families in Victoria for syndromes and isolated congenital HI [30]. For some couples, the value of understanding the prognosis, for example in non-syndromic isolated HI, could restore reproductive confidence despite knowing of a 25 or 50% recurrence risk.
A strength of this study was the integration of paediatric, otolaryngology and genetic services in a single clinic visit for families. This model was introduced specifically for this study and demonstrated benefit to both families and clinicians. Families had a single point of contact for booking the appointment and were only required to attend the service once to see all three specialties, and multidisciplinary communication was improved. The clinic model also resulted in deep phenotyping of cases which provided a more comprehensive interpretation of the data and can increase diagnostic yield [31] and contribute to the understanding of newly described genetic diseases. One participant had a variant in the SON gene identified which had recently been described as causing a complex developmental phenotype [32], consistent with this child’s features but in which HI is not described. As more cases are recognised this may become part of the phenotype associated with this gene. In a minority of cases, the molecular diagnosis identified did not provide a definitive explanation HI, yet was likely to have been causally related. For example the infant with SPTB variants and severe neonatal haemolytic anaemia was acutely unwell in the newborn period and required multiple blood transfusions and intravenous antibiotics prior to newborn hearing screening; haemolytic anaemia, antibiotics and admission to a neonatal intensive care unit are recognised factors that increase the risk of HI [33, 34].
The majority of cases were investigated for congenital CMV through the integrated clinic. This was primarily done on PCR of the newborn bloodspot card, except if the infant presented to clinic prior to 21 days of age when saliva PCR for CMV was performed. Five CMV positive cases were identified on newborn bloodspot card. This rate of detection, 5%, is comparable with the 6% when testing all infants who did not pass their initial hearing screening [35]. One of these cases also had pathogenic GJB2 compound heterozygous variants identified. The remaining four did not have any variants in GJB2. The number of cases of CMV was too small to support or refute the finding that the rate of heterozygous GJB2 variants is higher in infants with HI with congenital CMV than in those without [36].
Another strength of the study is the use of WES which provides the opportunity to re-analyse negative cases. Rapid progress in gene discovery requires targeted gene lists to be regularly updated. Since 2015 when the list of 144 genes applied to this cohort was designed there have been multiple gene discoveries. Review of the gene list and re-analysis of this cohort may further increase the diagnostic yield [37].
Microarray contributed to 11% (7/61) of diagnoses in this cohort. This rate is consistent with previously published data on the contribution of CNV to cases of HI [16]. The three STRC deletion cases required confirmation using MLPA due to the small size and the presence of a pseudogene which made it difficult to obtain supporting evidence from the WES data. These issues (detection of CNVs and accurate calling in the presence of pseudogenes) are likely to be overcome in the future with use of long-read technology or whole genome sequencing rather than WES.
Other technical challenges were difficulties and discrepancies interpreting variants utilising the ACMG guidelines. These issues are well-recognised [38] particularly in relation to the reporting of VUSs. The use of a VUS sub-classification system enables stratification of VUS variants, recognising those variants that are likely to be re-classified as LP or P in the future. For this reason the VUS-FP variants are included in this analysis.
The emergence of precision therapies in genetic medicine will increase the value of early molecular diagnosis. This is particularly relevant in HI syndromes where multiple senses are affected, resulting in significant disability in an otherwise normally developing child. An example of this is found in the participant with Stickler syndrome. A molecular diagnosis in the child has allowed for segregation in four clinically affected family members. This family can now access therapy to prevent retinal detachment [39] and therefore visual loss. In the future, gene editing and CRISPR/CAS-9 technology will expand the potential for further treatment in patients with a known genotype, particularly in organs such as the eye which are easily accessible. One of the more common variants causing Usher syndrome, USH2A c.2299delG, is the target of new trials into the treatment and prevention of retinal disease [40]. The participant in this cohort who has this variant, as well as the affected sibling, may now have the opportunity to participate in clinical trials aimed at preventing visual loss in Usher syndrome.
As sequencing becomes more common, knowledge about genotype–phenotype correlations is expanding. A case example is a novel homozygous nonsense variant in the COCH gene, c.1053C>A, that was identified in a non-dysmorphic infant with moderate bilateral sensorineural HI and healthy, hearing consanguineous parents. Heterozygous missense variants and in frame deletions in COCH have been reported as causing DFNA9—a dominant progressive hearing loss and vestibular dysfunction with adult onset [41], with the genetic mechanism postulated to be a dominant negative or gain-of-function effect rather than haploinsufficiency [42]. However, recent evidence suggests that bi-allelic loss-of-function variants result in congenital HI [43], consistent with the phenotype seen in our patient.
The point at which genetic testing is offered and the timeliness of results return are important factors in assessing the utility and economic impact of the technology [44]. Our data support what is already known about the prevalence of GJB2 as a common cause of congenital HI [45] and we suggest that testing for variants in this gene could precede genomic sequencing in the current resource-limited healthcare setting. A comprehensive economic evaluation is underway to ascertain the cost-effectiveness of this method of testing and the impact on the health system. Patient and parent preferences are also important to consider. Having a newborn with an unexpected diagnosis of HI is a stressful event and capacity to understand and consent for a complex test may be limited. The parents of 50 infants declined testing in this cohort and 18% of this group provided ‘feeling overwhelmed’ as their primary reason for declining, replicating the recruitment experience in other recent studies offering parents of newborns genomic sequencing [46].
This study highlights the benefits of early genetic diagnosis for infants with congenital HI. Molecular diagnosis in this cohort has resulted in streamlined and personalised screening protocols and added to a deepening understanding of genotype–phenotype correlations. It has also had the often under-represented value of providing an answer for families about why their child has HI.
This study has provided an understanding of the molecular basis of congenital HI in our population, which is the essential first step in understanding congenital HI and the basis upon which investigation, management, treatment and precision therapy can be developed. The integrated model of care provided in this study, of paediatric, genetic and otolaryngology services with comprehensive genetic testing demonstrated superior care for infants with HI. It resulted in higher diagnostic yield and streamlined management for both families and the healthcare system. This cohort has provided a real model on which to evaluate the utility and limitations of WES, and a framework for how genomic tests could be rolled out to complement newborn hearing screening.
Change history
09 November 2020
An amendment to this paper has been published and can be accessed via a link at the top of the paper.
References
Nishio SY, Usami S. Deafness gene variations in a 1120 nonsyndromic hearing loss cohort: molecular epidemiology and deafness mutation spectrum of patients in Japan. Ann Otol Rhinol Laryngol. 2015;124:49–60.
Sloan-Heggen CM, Bierer AO, Shearer AE, Kolbe DL, Nishimura CJ, Frees KL, et al. Comprehensive genetic testing in the clinical evaluation of 1119 patients with hearing loss. Hum Genet 2016;135:441–50.
Mehta D, Noon SE, Schwartz E, Wilkens A, Bedoukian EC, Scarano I, et al. Outcomes of evaluation and testing of 660 individuals with hearing loss in a pediatric genetics of hearing loss clinic. Am J Med Genet Part A. 2016;170:2523–30.
Bademci G, Foster J 2nd, Mahdieh N, Bonyadi M, Duman D, Cengiz FB, et al. Comprehensive analysis via exome sequencing uncovers genetic etiology in autosomal recessive nonsyndromic deafness in a large multiethnic cohort. [Erratum appears in Genet Med. 2016 Aug;18(8):859; PMID: 27494218]. Genet Med. 2016;18:364–71.
Zazo Seco C, Wesdorp M, Feenstra I, Pfundt R, Hehir-Kwa JY, Lelieveld SH, et al. The diagnostic yield of whole-exome sequencing targeting a gene panel for hearing impairment in The Netherlands. Eur J Hum Genet. 2017;25:308–14.
Likar T, Hasanhodzic M, Teran N, Maver A, Peterlin B, Writzl K. Diagnostic outcomes of exome sequencing in patients with syndromic or non-syndromic hearing loss. PLoS ONE. 2018;13:e0188578.
Talbi S, Bonnet C, Riahi Z, Boudjenah F, Dahmani M, Hardelin JP, et al. Genetic heterogeneity of congenital hearing impairment in Algerians from the Ghardaia province. Int J Pediatr Otorhinolaryngol. 2018;112:1–5.
Sheppard S, Biswas S, Li MH, Jayaraman V, Slack I, Romasko EJ, et al. Utility and limitations of exome sequencing as a genetic diagnostic tool for children with hearing loss. Genet Med. 2018:20:1663–76.
Cabanillas R, Dineiro M, Cifuentes GA, Castillo D, Pruneda PC, Alvarez R, et al. Comprehensive genomic diagnosis of non-syndromic and syndromic hereditary hearing loss in Spanish patients. BMC Med Genom. 2018;11:58.
Sakuma N, Moteki H, Takahashi M, Nishio SY, Arai Y, Yamashita Y, et al. An effective screening strategy for deafness in combination with a next-generation sequencing platform: a consecutive analysis. J Hum Genet. 2016;61:253–61.
Atik T, Onay H, Aykut A, Bademci G, Kirazli T, Tekin M, et al. Comprehensive analysis of deafness genes in families with autosomal recessive nonsyndromic hearing loss. PLoS ONE. 2015;10:e0142154.
Сhurbanov AY, Karafet TM, Morozov IV, Mikhalskaia VY, Zytsar MV, Bondar AA, et al. Whole Exome Sequencing Reveals Homozygous Mutations in RAI1, OTOF, and SLC26A4 Genes Associated with Nonsyndromic Hearing Loss in Altaian Families (South Siberia). PLoS ONE 2016;11:e0153841.
Shearer AE, Smith RJ. Massively parallel sequencing for genetic diagnosis of hearing loss: the new standard of care. Otolaryngol Head Neck Sur. 2015;153:175–82.
Sommen M, Wuyts W, Van Camp G. Molecular diagnostics for hereditary hearing loss in children. Expert Rev Mol Diagn. 2017;17:751–60.
Abou Tayoun AN, Al Turki SH, Oza AM, Bowser MJ, Hernandez AL, Funke BH, et al. Improving hearing loss gene testing: a systematic review of gene evidence toward more efficient next-generation sequencing-based diagnostic testing and interpretation. Genet Med. 2016;18:545–53.
Shearer AE, Kolbe DL, Azaiez H, Sloan CM, Frees KL, Weaver AE, et al. Copy number variants are a common cause of non-syndromic hearing loss. Genome Med. 2014;6:37.
Markova SP, Brozkova DS, Lassuthova P, Meszarosova A, Krutova M, Neupauerova J, et al. STRC gene mutations, mainly large deletions, are a very important cause of early-onset hereditary hearing loss in the Czech population. Genet Test Mol Biomark. 2018;22:127–34.
Bademci G, Diaz-Horta O, Guo S, Duman D, Van Booven D, Foster J 2nd, et al. Identification of copy number variants through whole-exome sequencing in autosomal recessive nonsyndromic hearing loss. Genet Testing Mol Biomark 2014;18:658–61.
Downie L, Halliday JL, Burt RA, Lunke S, Lynch E, Martyn M, et al. A protocol for whole-exome sequencing in newborns with congenital deafness: a prospective population-based cohort. BMJ Paediatr Open. 2017;1:e000119.
Mathers CS, A, Concha, M. Global burden of hearing loss in the year 2000. https://www.who.int/healthinfo/statistics/bod_hearingloss.pdf: World Health Organisation. 2000.
Sung V, Downie L, Paxton G, Liddle K, Birman C,WC, et al. Childhood Hearing Australasian Medical Professionals (CHAMP) network: consensus guidelines on investigation and clinical management of childhood hearing loss. J Paediatr Child Health. 2019;55;1013–22.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24.
McLaughlin HM, Ceyhan-Birsoy O, Christensen KD, Kohane IS, Krier J, Lane WJ, et al. A systematic approach to the reporting of medically relevant findings from whole genome sequencing. BMC Med Genet. 2014;15:134.
Australian Bureau of Statistics. Available from: https://www.abs.gov.au [26 August 2016].
van der Tuin K, Mensenkamp AR, Tops CMJ, Corssmit EPM, Dinjens WN, van de Horst-Schrivers ANA, et al. Clinical Aspects of SDHA-Related Pheochromocytoma and Paraganglioma: A Nationwide Study. J Clin Endocrinol Metab. 2018;103:438–45.
Splawski I, Tristani-Firuouzi M, Lehmann MH, Sanguinetti MC, Keating MT. Mutations in the hminK gene cause long QT syndrome and suppress IKs function. Nat Genet 1997;17:338–40.
Weeke P, Mosley JD, Hanna D, Delaney JT, Shaffer C, Wells QS, et al. Exome sequencing implicates an increased burden of rare potassium channel variants in the risk of drug-induced long QT interval syndrome. J Am Coll Cardiol. 2014;63:1430–7.
Wilson BT, Strong A, O’Kelly S, Munkley J, Stark Z. Metronidazole toxicity in Cockayne syndrome: a case series. Pediatrics 2015;136:e706–8.
Laugel V. Cockayne syndrome: the expanding clinical and mutational spectrum. Mech Ageing Dev. 2013;134:161–70.
Poulton A, Lewis S, Hui L, Halliday JL. Prenatal and preimplantation genetic diagnosis for single gene disorders: A population-based study from 1977 to 2016. Prenat Diagn 2018;38:904–10.
Stark Z, Tan TY, Chong B, Brett GR, Yap P, Walsh M, et al. A prospective evaluation of whole-exome sequencing as a first-tier molecular test in infants with suspected monogenic disorders. Genet Med. 2016;18:1090–6.
Kim JH, Shinde DN, Reijnders MRF, Hauser NS, Belmonte RL, Wilson GR, et al. De novo mutations in SON disrupt RNA splicing of genes essential for brain development and metabolism, causing an intellectual-disability syndrome. Am J Hum Genet. 2016;99:711–9.
Umehara T, Hosokawa S, Kita JY, Takahashi G, Okamura J, Nakanishi H, et al. Risk factors and prognostic factors of hearing impairment in neonatal intensive care unit-treated infants. Audiol Neuro-Otol. 2019;24:84–9.
Farrell AN, Landry AM, Yee ME, Leu RM, Goudy SL. Sensorineural hearing loss in children with sickle cell disease. Int J Pediatr Otorhinolaryngol. 2019;118:110–4.
Rawlinson WD, Palasanthiran P, Hall B, Al Yazidi L, Cannon MJ, Cottier C, et al. Neonates with congenital cytomegalovirus and hearing loss identified via the universal newborn hearing screening program. J Clin Virol. 2018;102:110–5.
Ross SA, Novak Z, Kumbla RA, Zhang K, Fowler KB, Boppana S. GJB2 and GJB6 mutations in children with congenital cytomegalovirus infection. Pediatr Res. 2007;61:687–91.
Stark Z, Schofield D, Martyn M, Rynehart L, Shrestha R, Alam K, et al. Does genomic sequencing early in the diagnostic trajectory make a difference? a follow-up study of clinical outcomes and cost-effectiveness. Genet Med. 2018;21:516.
Amendola LM, Jarvik GP, Leo MC, McLaughlin HM, Akkari Y, Amaral MD, et al. Performance of ACMG-AMP variant-interpretation guidelines among nine laboratories in the clinical sequencing exploratory research consortium. Am J Hum Genet. 2016;99:247.
Fincham GS, Pasea L, Carroll C, McNinch AM, Poulson AV, Richards AJ, et al. Prevention of retinal detachment in Stickler syndrome: the Cambridge prophylactic cryotherapy protocol. Ophthalmology 2014;121:1588–97.
Calzetti G, Levy RA, Cideciyan AV, Garafalo AV, Roman AJ, Sumaroka A, et al. Efficacy outcome measures for clinical trials of USH2A caused by the common c.2299delG mutation. Am J Ophthalmol. 2018;193:114–29.
Makishima T, Rodriguez CI, Robertson NG, Morton CC, Stewart CL, Griffith AJ. Targeted disruption of mouse Coch provides functional evidence that DFNA9 hearing loss is not a COCH haploinsufficiency disorder. Hum Genet 2005;118:29–34.
Masuda M, Mutai H, Arimoto Y, Nakano A, Matsunaga T. A novel frameshift variant of COCH supports the hypothesis that haploinsufficiency is not a cause of autosomal dominant nonsyndromic deafness 9. Biochem Biophys Res Commun. 2016;469:270–4.
Janssensdevarebeke SPF, Van Camp G, Peeters N, Elinck E, Widdershoven J, Cox T, et al. Bi-allelic inactivating variants in the COCH gene cause autosomal recessive prelingual hearing impairment. Eur J Hum Genet. 2018;26:587–91.
Dillon OJ, Lunke S, Stark Z, Yeung A, Thorne N, Gaff C, et al. Exome sequencing has higher diagnostic yield compared to simulated disease-specific panels in children with suspected monogenic disorders. Eur J Hum Genet. 2018;26:644–51.
Chan DK, Chang KW. GJB2-associated hearing loss: systematic review of worldwide prevalence, genotype, and auditory phenotype. Laryngoscope 2014;124:E34–53.
Genetti CA, Schwartz TS, Robinson JO, VanNoy GE, Petersen D, Pereira S, et al. Parental interest in genomic sequencing of newborns: enrollment experience from the BabySeq Project. Genet Med. 2019;21:622–30.
Acknowledgements
Ellie Prawer, Gemma Brett, Belinda Creighton, Samantha Ayres, Yana Smagarinsky, Jonathan Berg, Cynthia Powell, Libby Smith and the VicCHILD team, UsherKids Australia.
Funding
This work was funded by the Melbourne Genomics Health Alliance, supported by the Victorian State Government.
Author information
Authors and Affiliations
Consortia
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Downie, L., Halliday, J., Burt, R. et al. Exome sequencing in infants with congenital hearing impairment: a population-based cohort study. Eur J Hum Genet 28, 587–596 (2020). https://doi.org/10.1038/s41431-019-0553-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-019-0553-8
This article is cited by
-
PKHD1L1, a gene involved in the stereocilia coat, causes autosomal recessive nonsyndromic hearing loss
Human Genetics (2024)
-
Addition of an affected family member to a previously ascertained autosomal recessive nonsyndromic hearing loss pedigree and systematic phenotype-genotype analysis of splice-site variants in MYO15A
BMC Medical Genomics (2022)
-
AudioGene: refining the natural history of KCNQ4, GSDME, WFS1, and COCH-associated hearing loss
Human Genetics (2022)
-
Exome sequencing of families from Ghana reveals known and candidate hearing impairment genes
Communications Biology (2022)
-
Unraveling the genetic complexities of combined retinal dystrophy and hearing impairment
Human Genetics (2022)
