
Abstract
The posterior fossa of the cranium contains the cerebellum and brainstem. Processes that reduce the volume of the posterior fossa squeeze the cerebellum and brainstem caudally, resulting in Chiari I malformation (CM1). CM1 causes neck pain, balance issues, decreased motor skills and headaches in those affected. We have posterior fossa measurements and whole exome sequence data on individuals from 7 extended families from Russia that have a family history of CM1. We performed parametric linkage analyses using an autosomal dominant inheritance model with a disease allele frequency of 0.01 and a penetrance of 0.8 for carriers and 0.0 for non-carriers. Variant-based two-point linkage analysis and gene-based linkage analysis was performed. Our results found a genome-wide significant signal on chromosome 1q43-44 (max HLOD = 3.3) in the variant-based analysis and 12q23 (max HLOD = 4.2) in the gene-based analysis. In both cases, the signal was driven by a single (different) family that contained a long, linked haplotype across the region in question. Using functional annotation, we were able to identify several rare nonsynonymous variants that were enriched in each family. The best candidate genes were rs765865412:G>A in MYBPC1 for the 12q haplotype and rs61749963:A>G in COX20 for the 1q haplotype. Good candidate variants in the 1q haplotype were also identified in CEP170 and AKT. Further laboratory work is planned to verify the causality of these genes.
Similar content being viewed by others


Introduction
Chiari I malformation (CM1) is an anatomical deformity characterized by inferior positioning of the cerebellar tonsils below the foramen magnum. Underdevelopment of the posterior fossa is often associated with CM1 [1,2,3,4,5]. Clinical symptoms arising from the malformation are variable, including headache, usually occipital and worsened by cough or Valsalva maneuver, neck pain, and poor balance. Many individuals with CM do not show symptoms and are diagnosed by brain or cervical spine MRI while being evaluated for another disorder. In CM1, the cerebellar tonsils extend ≥5 mm below the foramen magnum [1] (Fig. 1a). The role of genetics in the development of CM1 is suspected but no risk variants for CM1 has been found, except for those associated with a syndrome, such as achondroplasia. Milhorat et al. studied a cohort of 364 patients with CM1 and found that 43 patients (12%) reported positive family histories of CM1 or syringomyelia, a disorder associated with CM [1]. Pedigree analysis was performed in 21 of the 43 patients and was consistent with autosomal dominant or recessive patterns. Others have shown that CM1 results from the posterior fossa being smaller than normal in the majority of cases [2,3,4] and of insufficient size to contain the cerebellum and brainstem [5], rather than being the result of a primary abnormality of the cerebellum. The hindbrain deformity in CM1 resolves after surgical craniocervical decompression and duraplasty which enlarges the posterior fossa. Following this procedure, the cerebellar tonsils ascend into the expanded posterior fossa and revert from a pointed shape to a normal, rounded morphology [6, 7] suggesting that the abnormal shape of the cerebellar tonsils seen in CM1 is acquired by tonsillar impaction in the foramen magnum and the upper portion of the spinal canal, rather than resulting from a congenital abnormality of the tonsils [7]. These studies suggest that genetic factors leading to underdevelopment of the posterior cranial fossa are responsible for CM1 development. By considering affection status to be based on the primary problem of posterior fossa underdevelopment, rather than by the presence of CM1 alone, the power to detect an underlying, heritable factor may increase.

CM1 and small posterior fossa phenotyping. a CM1 phenotyping midsagittal T1-weighted MR-image showing the cerebellar tonsils (white arrow) extending ≥5 mm below the foramen magnum in a patient with CM1. b Small posterior fossa diagram of the measurements taken in the midsagittal plane, adapted from an earlier, non-copyrighted publication: Heiss JD, Suffredini G, Bakhtian KD, Sarntinoranont M, Oldfield EH. Normalization of hindbrain morphology after decompression of Chiari malformation Type I. J Neurosurg. 2012;117(5):942-6. Measurements (mm) to assess for the small bone phenotype included: supraocciput length (so, internal occipital protuberance to opisthion); clivus length (cl, apex of dorsum sellae to basion); and basiocciput length (bo, the portion of the clivus below the sphenoocipital synchondrosis). Cerebellar tonsillar ectopia (T, maximum extension of the cerebellar tonsils caudal to the foramen magnum) was not used to assess for the small bone phenotype
There have been few genetic studies on CM1, presumably because of the small number of family members (2–3) affected in each established pedigree [1,2,3,4]. Boyles et al. identified significant linkage to 15q21.1-22.3 and found posterior fossa volume to be highly heritable in 23 families [8]. Further studies have confirmed the heritability of posterior fossa volume [9] and found suggestive linkage to loci at 8q, 22q, 1q, and 12p [9, 10]. A recent filtration approach identified shared variants in two affected families [11]. The lone association study found no significant variants [12].
The formation of the upper and lower skull occurs by two distinct processes. In the supratentorial skull, intramembranous bone growth takes place, allowing the supratentorial skull to expand in response to the underlying pressure of the growing brain. The posterior fossa develops from endochondral (within cartilage) bone growth of the occipital sclerotomes (O1–O4), which are embryologic spinal elements [13]. The ring of bone formed by the ossification centers at the foramen magnum and lower posterior fossa develops in the same fashion as the vertebral body and arch. The diameter and height of the ring that forms around the neural elements of the posterior fossa depends on the amount and shape of the bone that is produced at each ossification center. Ossification in the lower part of the posterior fossa is genetically determined, being unaffected by brain volume and pressure, which expands the supratentorial skull to its adult volume. Thus, genetic disorders can result in disparities in growth and volume of the supra- and infratentorial parts of the skull [14]. For example, achondroplasia is caused by an autosomal dominant variant in FGFR3 gene that results in dwarfism and impaired development of the bones of the skull base, but does not reduce the size of the supratentorial skull [15]; the small posterior fossa results in herniation of the cerebellar tonsils identical to what is seen in CM1 [16]. Patients with idiopathic CM1 are not dwarfs and the bone dysplasia is restricted to the skull base in CM1, so the variant affecting function must be expressed primarily in the skull base [17, 18].
Patients with CM1 often have syringomyelia, which affects about 21,000 Americans [19]. The myelopathy from syringomyelia usually develops during the 2nd–5th decades of life and causes symptoms of paralysis, sensory loss, and chronic pain. The natural history of syringomyelia is typically one of gradual, stepwise neurologic deterioration over many years [20]. Finding a genetic locus for CM1 may lead to a better understanding of the etiology of CM1, which may lead to ways to prevent it and its associated condition syringomyelia from occurring, or to treat it earlier, sparing patients from its symptoms and neurologic dysfunction.
In Republic of Tatarstan in the Russian Federation a regional disease cluster with a high prevalence (413:100,000) of CM1 was found. In that cluster, about one-third of affected patients had an affected first-degree relative [21]. In collaboration with Dr. E. I. Bogdanov, Head of the Department of Neurology in Kazan State Medical University, Republic of Tatarstan of Russia, we identified seven extended families with at least five members affected by CM1 and a small posterior fossa. The probability of finding a genetic locus harboring risk variants for these phenotypes increases in families with many members affected over many generations.
Methods
Recruitment and study design
Eligible participants for this study were required to (1) have CM1 and a family member with syringomyelia or CM1 or (2) be a family member of a patient with CM1 and have at least two immediate family members diagnosed with CM1. Adults and minors were eligible for this study. Patients were excluded if they had a contraindication to MRI scanning, were unable to understand the risks of testing, were under one year of age, or could not undergo MRI scanning without sedation. Recruitment was through self- and physician referral. More information about the study can be found at ClinicalTrials.gov using identifier NCT0004738. All subjects provided informed consent and the protocols adhere to the Declaration of Helsinki and were approved by the institutional review boards of the NINDS and NHGRI. The average age of participants was 38.52 years with a standard deviation of 3.17.
Small posterior fossa phenotype determination
The small posterior fossa phenotype was established if the posterior fossa volume/supratentorial volume < 0.15. This method assessed if the posterior fossa volume was small compared to supratentorial volume [14]. The small posterior fossa phenotype was also established by the clivus length ≤40 mm and basiocciput length (the portion of the clivus below the sphenooccipital synchondrosis) ≤21 mm, and the length of the supraocciput (internal occipital protuberance to the opisthion) ≤38 mm (Fig. 1b). Earlier studies have supported the presence of clival, basioccipital, and supraoccipital hypoplasia in patients with CM1 [1,2,3,4,5, 22]. Threshold values were based upon average values from healthy normal individuals. These measurements were not found to correlate with age (Supplementary Figs. 1, 2). All measurements are given in Supplementary Table 1.
Participants had an outpatient protocol visit. Participants attended appointments for a history, physical, and neurological examination, blood draw, and MR-imaging of the brain and cervical spine. Family pedigrees for CM1 and for small posterior fossa were established. For linkage analysis, DNA for each patient was extracted from blood lymphocytes contained in a 10 ml blood sample, stored, and sent for whole exome sequencing (WES).
MR-imaging
Study participants underwent SPGR (spoiled gradient recalled) and T1-weighted MR-imaging of the brain and cervical spinal cord to assess the length of the posterior fossa bones, the presence of CM1 (defined as the inferior aspect of the cerebellar tonsils lying ≥5 mm caudal to the foramen magnum), and the presence of syringomyelia. Intravenous contrast was not used.
Whole exome sequencing and quality control
WES was performed on 62 samples at the National Intramural Sequencing Center (NISC). Variants with a depth (DP) and genotype quality (GQ) score of less than 10 were removed, as were all monomorphic variants. PLINK [23] was used for additional quality control. Markers with more than 80% missingness and individuals with more than 5% missingness were removed. PLINK was also used to check Mendelian inconsistencies and identity-by-descent (IBD) values. Variants with Mendelian inconsistencies in a single family were set to zero in that family only, while variants with Mendelian inconsistencies across multiple families were removed from all families. One individual was removed as he was found to be unrelated to both of his parents. All unrelated individuals had IBD values consistent with being unrelated. Thirty-three ungenotyped people were added into the data set to ensure proper familial relationships. These are individuals that we know existed through family history, but who were missing phenotype and genotype information because they were either deceased or were unwilling or unable to participate in the study. All genotype information was set to missing for these individuals; phenotype information for these individuals was used if present, otherwise phenotype information was also set to missing. After quality control, the data set consisted of 401,733 variants (both SNVs and indels) from 95 people (62 genotyped) from 7 extended families. Dataset level allele frequencies were then calculated by sib-pair [24]. Genotype and phenotype files are available for download on dbGaP (https://www.ncbi.nlm.nih.gov/gap). The study ID is 32578, the accession number is phs001795.v1.p1, and the title is “Genetic Analysis of the Chiari I Malformation”.
Parametric linkage analysis and functional annotation of variants
All parametric linkage analyses assumed an autosomal dominant mode of transmission, assuming a disease allele frequency of 1% with an 80% penetrance for carriers and 0% phenocopy rate. Sensitivity analyses were also performed using a phenocopy rate of 0.005; no significant differences in LOD scores were observed between the two models. We performed both variant-based and gene-based genetic linkage. Analysis was performed on two discrete phenotypes, small posterior fossa and CM1 diagnosis. Both phenotypes were binary, with individuals being labeled as an affected case, unaffected control, or unknown. For small posterior fossa, there were 46 genotyped cases. For CM1, there were 31 genotyped cases.
The variant-based linkage analysis tested for linkage between each SNV and the designated phenotype and was performed using the TwoPointLods [25]. Gene-based analysis was performed by creating haplotypes corresponding to particular genes and performing two-point linkage on the genic haplotypes. We used the collapsed haplotype pattern (CHP) method, implemented through the SEQLinkage [26], to create the gene-based haplotypes. This method uses rare variants (defined as MAF < 0.05) to create short regional haplotypes that correspond to genes (determined by RefSEQ). The gene-based haplotypes essentially function as multiallelic pseudo-markers in the two-point linkage analysis. Two-point linkage analysis was performed on the pseudo-markers using MERLIN [27]. Variants were annotated with wANNOVAR [28, 29] using hg19.
Results
HLOD scores for small posterior fossa and CM1 affection
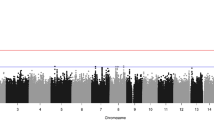
Variant-based analysis identified two genome-wide significant SNVs for the small posterior fossa phenotype, one located in the exon of OR2T8 (HLOD = 3.3) at 1q44 and the second located the UTR of CHML (HLOD = 3.3) at 1q43 (Fig. 2a). Here, we use the Lander and Kruglyak values of (H)LOD ≥ 3.3 and (H)LOD ≥ 1.9 as the genome-wide significant and suggestive thresholds [30]. Six additional variants had LOD scores near significance (HLOD ≥ 3.0). All of these variants were located at 1q43-44 except for one, which was located in an intron of ANO4 (HLOD = 3.0) at 12q23.1. In total, there were 350 suggestive variants, with 84 being located at 1q43-44 and 33 located at 12q23-24.11 (Supplementary Table 2).

Genome-wide HLOD Scores for Small Posterior Fossa. The genome-wide HLOD scores combined across all 7 families for the variant-based (a) and the gene-based (b) linkage analyses. The lines at 3.3 and 1.9 represent the significant and suggestive thresholds as recommended by Lander and Kruglyak
There were 18 genome-wide significant genes in the gene-based linkage analysis (Fig. 2b). Four of these genes were located in the 12q23-24 region identified as significant in the variant-based analysis, the highest of any chromosomal region. The genes were MYBPC1 (HLOD = 4.2) at 12q23.2, GNPTAB (HLOD = 3.5) at 12q23.2, CMKLR1 (HLOD = 3.3) at 12q23.3 and HECTD4 (HLOD = 3.4) at 12q24.13. The significant signal at 1q found in the variant-based analysis was still present, though it had slightly decreased, with the highest signal in the region located on the TSNAX-DISC1 (HLOD = 3.2) at 1q42.2. The highest overall HLOD score in the gene-based analysis was located on EPS15 (HLOD = 4.9) at 1p32.3 (Supplementary Table 3).
Neither the variant-based analyses nor the gene-based analyses identified any genome-wide significant signals when using CM1 affection as the phenotype, though multiple suggestive signals were identified in both analyses (Supplementary Fig. 3).
Individual family LOD scores
The moderately large size of the families used in this study allowed us to powerfully determine which chromosomal region was linked to the phenotype in each family by examining the individual family LOD scores closely. We were interested in whether any of the significant signals were being driven primarily by a single family or if any signal was extremely high within a family. This would imply that a particular family was harboring a causal gene of large effect in that region.
We observed that the significant signal on 1q was being driven by a single family. Family 4 had a large linkage peak at 1q43-44 in both the variant-based (Fig. 3a) and gene-based analyses (Supplementary Fig. 4a). Family 4 is a 4 generational family with 7 genotyped cases and 16 genotyped people overall. Though none of the individual LOD scores were genome-wide significant (unlikely in a single moderate-sized family) there were 77 suggestive variants and 8 suggestive genes located within 1q43-44 in this family. The highest overall LOD scores were 2.9 in the variant based analysis (shared by 10 SNVs) and 2.8 in the gene-based analysis (shared by 4 genes). The full list of suggestive genes can be found in Table 1. A selected list of variants can be viewed in Table 2; the full list is available in Supplementary Table 4. Plots of chromosome 1 (Fig. 3b, c) for both sets of analyses in family 4 reveal a linked haplotype across the 1q43–1q44 region at approximately the 2.9 LOD mark in the variant-based analyses and 2.8 in the gene-based analyses. There is almost no negative signal underneath these haplotypes, while the rest of the chromosome always contains negative LOD scores even in the presence of linked variants or haplotypes (e.g., the region with some variants/genes showing LODs of ~1 on 1p at ~50,000,000 bp). This is an excellent indication that the haplotype at 1q43-44 is strongly linked to the trait and that the causal variant in this region may have a large effect on the trait.

LOD scores for Family 4. This figure shows the genome-wide LOD scores for family 4 for the variant based linkage analysis (a) as well as zoomed plots of the chromosome 1 LOD scores for family 4 for the variant-based (b) and gene-based (c) linkage analyses. The line at 1.9 represents the suggestive threshold as recommended by Lander and Kruglyak
We observe a similar scenario with family 22, a three generational family with 8 genotyped cases and 13 genotyped individuals. Both the variant-based (Fig. 4a) and the gene-based (Supplementary Fig. 4b) linkage analyses showed a large peak at 12q23-24.11. This is the primary driver of the significant and highly suggestive linkage peaks found at that region in the overall analyses. The gene-based analysis identified seven suggestive genes with a LOD score of 2.2 in the chromosomal region (Table 3), while the variant-based analysis found 27 suggestive variants that shared the top LOD score of 2.2 in the family and an additional 61 suggestive variants within the region (Table 4 and Supplementary Table 5). The plots of chromosome 12 for both analyses reveal a long, linked haplotype across the 12q23-24.11 region (Fig. 4b, c). This haplotype is longer than the one identified at 1q43-44 in family 4. Like the 1q haplotype, this haplotype also has almost no negative LOD scores underneath it, suggestive of being highly linked to the phenotype.

LOD scores for Family 22. This figure shows the genome-wide LOD scores for family 22 for the variant based linkage analysis (a) as well as zoomed plots of the chromosome 12 LOD scores for family 4 for the variant-based (b) and gene-based (c) linkage analyses. The line at 1.9 represents the suggestive threshold as recommended by Lander and Kruglyak
Functional annotation and cosegregation of candidate causal variants
Functional annotation was used to find any potential causal variants along the two linked haplotypes in families 4 and 22. The most promising variant along the 12q haplotype was rs765865412:G>A (hg19 chr12:g.102055019G>A), located in the MYBPC1 and was predicted to be possibly damaging. The minor allele is extremely rare; it is not in any 1000Genomes population and has a MAF of 0.00002 in non-Finnish Europeans in the Exome Aggregation Consortium (ExAC). The minor allele appears in our data set 8 times, in the 8 cases in family 22. It does not appear in any of the controls or unknowns outside the family. rs765865412:G>A variant is an excellent candidate for the causal variant along the 12q23-24.11 haplotype (Supplementary Fig. 5). The synonymous variant rs199921889:A>C (hg19 chr12:g.102158766A>C) (MAF < 0.001 in 1000Genomes) in GNPTAB also completely cosegregates with all family 22 cases.
Family 4 contained 14 nonsynonymous exonic variants across the haplotype on 1q. The best candidate nonsynonymous rare variant is rs61749963:A>G (hg19 chr1: g.245006474A>G) in COX20, which has a MAF in 1000Genomes Europeans of 0.012. The minor allele only appears once in each of the seven cases in family 4 and once in one unknown phenotype individual and nowhere else in the dataset. It is not predicted damaging (Supplementary Fig. 5). The intronic variant rs1901574:C>T (hg19 chr1:g.243335862C>T) in CEP170 (MAF 0.007) also segregates completely with those same individuals and thus is also an excellent candidate for causality. We note that within both pedigrees, there are no unaffected individuals that are carrying the best candidate disease linked variant allele and there are no phenocopies, i.e., all known affecteds carry the disease linked variant allele.
We did briefly perform a CNV analysis using CNVnator [31] to call CNVs along the haplotypes. We did not find any shared CNVs along the haplotype amongst affected individuals.
Discussion
CM1 has not been well analyzed by genetic linkage or association analyses [8,9,10,11], thus few risk loci have been identified. Our study was the first to perform linkage analysis on CM1 affected families using WES data. We identified two novel linked haplotypes at 1q43-44 and 12q23-24.11 for small posterior fossa, one of the probable underlying causes of CM1. Indeed, the vast majority (83%) of CM1 affected individuals had the small posterior fossa phenotype. The linked haplotypes were significantly higher than other signals within the two families and were characterized by very little to no negative signal underneath. These results led us to believe the haplotypes are harboring causal variant(s) of large effect. This is a highly significant finding for a rare disease with few identified risk loci.
Long linked haplotypes are expected in a linkage study. Unlike population-based studies, in which countless meioses have broken apart haplotypes, there are only a limited number of recombination events that can occur within small families. The result is longer haplotypes such as the ones identified in this study. However, this makes it more difficult to elucidate the causal variant.
We performed functional annotation to identify any potential causal variants located on the linked haplotypes. Since the small posterior fossa phenotype is rare and the gene-based tests (which only used variants with MAF < 0.05) recapitulated the linked haplotypes found in the variant-based analysis, we believe it likely the causal variant are rare (MAF ≤ 0.01). We were able to find several good, rare, candidate variants on both of the linked haplotypes. The most promising causal variant for the 12q23-24.11 haplotype is rs765865412:G>A, located in MYBPC1 gene at 12q23.2. The variant and gene have the highest overall LOD scores for family 22 in the variant-based and gene-based analyses (LOD = 2.18) and is genome-wide significant (HLOD = 4.15) across the families in the gene-based analysis. Recall that the variant is nonsynonymous exonic, predicted damaging by some databases, and extremely rare (ExAC MAF = 0.00002 in Europeans). In our data set, the minor allele only appears in the eight cases in Family 22 and nowhere else.
The MYBPC family encodes proteins expressed in striated muscles and MYBPC1 in particular encodes the slow skeletal isoform and plays an important role in muscle contraction [32]. MYBPC1 causes distal arthogryposis (DA) type 1 [33], a muscular skeletal disorder characterized by deformities of the hands and feet (e.g., permanently bent fingers/toes) and DA type 2 [34], where patients often display craniofacial abnormalities in addition to the hand/feet defects. Morpholino-mediated knockout of MYBPC1 in zebrafish was reported to cause poor growth of the head and eyes [35].
rs765865412:G>A is not one of the missense variants known to cause DA [33, 34]. rs765865412:G>A results in a substitution of aspartic acid to asparagine; this is different from the variants known to affect function [32]. It has been shown that multiple variants within MYBPC1 result in phenotypes of varying severity, so it is possible that this variant results in less severe phenotype. Mechanistically, one can speculate that perhaps this variant causes less severe muscle contraction and restricts growth at the base of the skull (resulting in a small posterior fossa) but does not result in the contractures of the hands and feet. Future laboratory work will be needed to confirm any molecular conjecture regarding the role of MYBPC1 in CM1. Two other rare variants of interest should be noted. rs199921889:A>C showed complete segregation in all family 22 cases and is located in GNPTAB, which has been implicated as causal in prenatal skeletal dysplasia [36]. rs7303227:C>A (hg19 chr12:g. 108984751C>A) in the 3′ UTR of TMEM119, is a gene associated with osteoblast differentiation [37], a relevant potential causal gene.
The second linked haplotype was located along 1q43-44 haplotype in family 4. The 1q43-44 telomere is the site of well-reported deletions/microdeletions that result in severe neurological/skeletal phenotypes such as microcephaly, structural disorders of the brain and abnormalities of the hands/feet [38,39,40]. The majority of these 1q43-44 deletions are de novo [41] and thus not likely to be responsible for the signals seen here. However, many of the genes located along the linked haplotype have been implicated as causal in the various neurological and skull structure phenotypes including AKT3 and COX20 [41, 42]. Deletions of AKT3, a serine/threonine protein kinase expressed in the adult brain have been implicated in microcephaly [41]. Three variants in the UTR of AKT3 were located along the linked haplotype in the variant-based analyses. Since the variants had a common MAF (~0.3), this gene’s signal was not reproduced in the gene-based analysis, though the gene remains a strong candidate for causality. One of the more promising rare variants along the haplotype was rs61749963:A>G in COX20. This variant had the highest LOD score (2.9) in the family and the COX20 gene had the highest overall score in the gene-based analysis. Though rs61749963:A>G was not predicted damaging, it is nonsynonymous exonic and rare (MAF = 0.012) in the general European population. The minor allele only appears in our data set in the seven cases (and one unknown) in family 4. COX20 is involved in the assembly of cytochrome C oxidase, a key component of mitochondrial respiration and a different variant has been implicated as causal for cerebellar ataxia and muscle hypotonia [43]. It is conceivable that a milder version of the phenotype could result in the small posterior fossa. Another strong potential causal variant is rs1901574:C>T in CEP170. Though the variant is intronic, it is rarer than COX20 (MAF = 0.007 in Europeans) and also shows complete cosegregation with all cases in family 4 and does not appear in any other families. CEP170 has been implicated in microcephaly [44]. In general, we note that there are multiple, potential causal variants located along the 1q haplotype. The variants in COX20, CEP170, and AKT look like strong candidates, but it is certainly possible the causal variant(s) are located elsewhere along the haplotype.
This study used WES genotypes in families to identify two strongly linked novel haplotypes (each in a different single family) for small posterior fossa, a potential underlying cause for CM1. The haplotypes were located at 1q43-44 and 12q23-24.11. This is the first time linked haplotypes have been identified for small posterior fossa using WES data. Both haplotypes spanned large genomic regions, and thus incorporated a large number of good candidate genes. Given the rarity of the trait, we believe that the causal variant is likely rare and we used functional annotation to identify potential rare causal variants along the haplotypes. We have identified rs765865412:G>A in MYBPC1 at 12q23.2 and rs61749963:A>G in COX20 at 1q44 as the best potential variants for causality. However, there are other strong causal variant candidates, especially along the 1q haplotype, with rs1901574:C>T in CEP170 and three variants in AKT being of particular interest due to their known roles in microcephaly. It is also possible that the true causal variants lie elsewhere along the haplotypes, perhaps even in the noncoding regions not sequenced here. Only functional studies will be able to determine causality and we initially plan functional studies on our best candidate genes: MYBPC1 on 12q and COX20, CEP170, and AKT on 1q to define their possible roles in determining the volume of the posterior fossa.
References
Milhorat TH, Chou MW, Trinidad EM, Kula RW, Mandell M, Wolpert C, et al. Chiari I malformation redefined: clinical and radiographic findings for 364 symptomatic patients. Neurosurgery. 1999;44:1005–17.
Stovner LJ, Bergan U, Nilsen G, Sjaastad O. Posterior cranial fossa dimensions in the Chiari I malformation: relation to pathogenesis and clinical presentation. Neuroradiology. 1993;35:113–8.
Nyland H, Krogness KG. Size of posterior fossa in Chiari type 1 malformation in adults. Acta Neurochir. 1978;40:233–42.
Vega A, Quintana F, Berciano J. Basichondrocranium anomalies in adult Chiari type I malformation: a morphometric study. J Neurol Sci. 1990;99:137–45.
Nishikawa M, Sakamoto H, Hakuba A, Nakanishi N, Inoue Y. Pathogenesis of Chiari malformation: a morphometric study of the posterior cranial fossa. J Neurosurg. 1997;86:40–7.
Barkovich AJ, Wippold FJ, Sherman JL, Citrin CM. Significance of cerebellar tonsillar position on MR. Am J Neuroradiol. 1986;7:795–9.
Heiss JD, Patronas N, DeVroom HL, Shawker T, Ennis R, Kammerer W, et al. Elucidating the pathophysiology of syringomyelia. J Neurosurg. 1999;91:553–62.
Boyles AL, Enterline DS, Hammock PH, Siegel DG, Slifer SH, Mehltretter L, et al. Phenotypic definition of Chiari type I malformation coupled with high-density SNP genome screen shows significant evidence for linkage to regions on chromosomes 9 and 15. Am J Med Genet Part A. 2006;140:2776–85.
Markunas CA, Enterline DS, Dunlap K, Soldano K, Cope H, Stajich J, et al. Genetic evaluation and application of posterior cranial fossa traits as endophenotypes for Chiari type I malformation. Ann Hum Genet. 2014;78:1–12.
Markunas CA, Soldano K, Dunlap K, Cope H, Asiimwe E, Stajich J, et al. Stratified whole genome linkage analysis of Chiari type I malformation implicates known Klippel-Feil syndrome genes as putative disease candidates. PLoS One. 2013;8:e61521.
Merello E, Tattini L, Magi A, Accogli A, Piatelli G, Pavanello M, et al. Exome sequencing of two Italian pedigrees with non-isolated Chiari malformation type I reveals candidate genes for cranio-facial development. Eur J Hum Genet. 2017;25:952–9.
Urbizu A, Toma C, Poca MA, Sahuquillo J, Cuenca-Leon E, Cormand B, et al. Chiari malformation type I: a case–control association study of 58 developmental genes. PLoS One. 2013;8:e57241.
Parke WW. Development of the spine. In: W.B. Saunders Co, editor. The Spine. Philadelphia: Rothman-Simeone; 1999. p. 3–27.
Badie B, Mendoza D, Batzdorf U. Posterior fossa volume and response to suboccipital decompression in patients with Chiari I malformation. Neurosurgery. 1995;37:214–8.
Rousseau F, Bonaventure J, Legeai-Mallet L, Pelet A, Rozet JM, Maroteaux P, et al. Mutations in the gene encoding fibroblast growth factor receptor-3 in achondroplasia. Nature. 1994;371:252–4.
Uematsu S, Wang H, Kopits SE, Hurko O. Total craniospinal decompression in achondroplastic stenosis. Neurosurgery. 1994;35:250–7.
Coria F, Quintana F, Rebollo M, Combarros O, Berciano J. Occipital dysplasia and Chiari type I deformity in a family. Clinical and radiological study of three generations. J Neurol Sci. 1983;62:147–58.
Stovner LJ, Cappelen J, Nilsen G, Sjaastad O. The Chiari type I malformation in two monozygotic twins and first-degree relatives. Ann Neurol. 1992;31:220–2.
Brewis M, Poskanzer DC, Rolland C, Miller H. Neurological disease in an English city. Acta Neurol Scand. 1966;42:1–89.
Boman K, Iivanainen M. Prognosis of syringomyelia. Acta Neurol Scand. 1967;43:61–8.
Bogdanov EI, Faizutdinova AT, Mendelevich EG, Sozinov AS, Heiss JD. Epidemiology of symptomatic chiari malformation in tatarstan: regional and ethnic differences in prevalence. Neurosurgery. 2019;84:1090–7.
Noudel R, Jovenin N, Eap C, Scherpereel B, Pierot L, Rousseaux P. Incidence of basioccipital hypoplasia in Chiari malformation type I: comparative morphometric study of the posterior cranial fossa. J Neurosurg. 2009;111:1046–52.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–75.
Duffy D SIB-PAIR: A program for simple genetic analysis v1.00.beta. 2008.
Thomas A. TwoPointsLods. 2017. http://www-genepi.med.utah.edu/~alun/software/docs/TwoPointLods.html.
Wang GT, Zhang D, Li B, Dai H, Leal SM. Collapsed haplotype pattern method for linkage analysis of next-generation sequence data. Eur J Hum Genet. 2015;23:1739–43.
Abecasis GR, Cherny SS, Cookson WO, Cardon LR. Merlin-rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet. 2002;30:97–101.
Chang X, Wang K. wANNOVAR: annotating genetic variants for personal genomes via the web. J Med Genet. 2012;49:433–6.
Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164.
Lander E, Kruglyak L. Genetic dissection of complex traits: guidelines for interpreting and reporting linkage results. Nat Genet. 1995;11:241–7.
Abyzov A, Urban AE, Snyder M, Gerstein M. CNVnator: an approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res. 2011;21:974–84.
Geist J, Kontrogianni-Konstantopoulos A. MYBPC1, an emerging myopathic gene: what we know and what we need to learn. Front Physiol. 2016;7:410.
Gurnett CA, Desruisseau DM, McCall K, Choi R, Meyer ZI, Talerico M, et al. Myosin binding protein C1: a novel gene for autosomal dominant distal arthrogryposis type 1. Hum Mol Genet. 2010;19:1165–73.
Li X, Zhong B, Han W, Zhao N, Liu W, Sui Y, et al. Two novel mutations in myosin binding protein C slow causing distal arthrogryposis type 2 in two large Han Chinese families may suggest important functional role of immunoglobulin domain C2. PLoS One. 2015;10:e0117158.
Ha K, Buchan JG, Alvarado DM, McCall K, Vydyanath A, Luther PK, et al. MYBPC1 mutations impair skeletal muscle function in zebrafish models of arthrogryposis. Hum Mol Genet. 2013;22:4967–77.
Aggarwal S, Coutinho MF, Dalal AB, Mohamed Nurul Jain SJ, Prata MJ, Alves S. Prenatal skeletal dysplasia phenotype in severe MLII alpha/beta with novel GNPTAB mutation. Gene. 2014;542:266–8.
Kanamoto T, Mizuhashi K, Terada K, Minami T, Yoshikawa H, Furukawa T. Isolation and characterization of a novel plasma membrane protein, osteoblast induction factor (obif), associated with osteoblast differentiation. BMC Dev Biol. 2009;9:70.
Mankinen CB, Sears JW, Alvarez VR. Terminal (1)(q43) long-arm deletion of chromosome no. 1 in a three-year-old female. Birth Defects Orig Artic Ser. 1976;12:131–6.
Boland E, Clayton-Smith J, Woo VG, McKee S, Manson FD, Medne L, et al. Mapping of deletion and translocation breakpoints in 1q44 implicates the serine/threonine kinase AKT3 in postnatal microcephaly and agenesis of the corpus callosum. Am J Hum Genet. 2007;81:292–303.
Hill AD, Chang BS, Hill RS, Garraway LA, Bodell A, Sellers WR, et al. A 2-Mb critical region implicated in the microcephaly associated with terminal 1q deletion syndrome. Am J Med Genet Part A. 2007;143A:1692–8.
Ballif BC, Rosenfeld JA, Traylor R, Theisen A, Bader PI, Ladda RL, et al. High-resolution array CGH defines critical regions and candidate genes for microcephaly, abnormalities of the corpus callosum, and seizure phenotypes in patients with microdeletions of 1q43q44. Hum Genet. 2012;131:145–56.
Gai D, Haan E, Scholar M, Nicholl J, Yu S. Phenotypes of AKT3 deletion: a case report and literature review. Am J Med Genet Part A. 2015;167A:174–9.
Szklarczyk R, Wanschers BF, Nijtmans LG, Rodenburg RJ, Zschocke J, Dikow N, et al. A mutation in the FAM36A gene, the human ortholog of COX20, impairs cytochrome c oxidase assembly and is associated with ataxia and muscle hypotonia. Hum Mol Genet. 2013;22:656–67.
Hemming IA, Forrest AR, Shipman P, Woodward KJ, Walsh P, Ravine DG, et al. Reinforcing the association between distal 1q CNVs and structural brain disorder: a case of a complex 1q43-q44 CNV and a review of the literature. Am J Med Genet Part B. 2016;171B:458–67.
Acknowledgements
The authors thank all study participants and their families. This work was funded in part by the Intramural Research Programs of the National Institute for Neurological Disorders and Stroke and the National Human Genome Research Institute of the National Institutes of Health. This work utilized the computational resources of the NIH HPC Biowulf cluster (http://hpc.nih.gov).
Funding
This work was funded in part by the Intramural Research Programs of the National Institute for Neurological Disorders and Stroke and the National Human Genome Research Institute of the National Institutes of Health.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Musolf, A.M., Ho, W.S.C., Long, K.A. et al. Small posterior fossa in Chiari I malformation affected families is significantly linked to 1q43-44 and 12q23-24.11 using whole exome sequencing. Eur J Hum Genet 27, 1599–1610 (2019). https://doi.org/10.1038/s41431-019-0457-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-019-0457-7
This article is cited by
-
Posterior cranial fossa and cervical spine morphometric abnormalities in symptomatic Chiari type 0 and Chiari type 1 malformation patients with and without syringomyelia
Acta Neurochirurgica (2021)
-
Functional and morphological changes in hypoplasic posterior fossa
Child's Nervous System (2021)
