
Abstract
Genome-wide association studies have led in the past to the discovery of susceptibility genes for many diseases including cancer and inflammatory conditions. However, a number of these studies did not realise their full potential. A first critical step in developing such large-scale studies is the choice of genotyping array with respect to the study goal. Coverage is the central criterion for array evaluation. We distinguish between estimates of global coverage across the genome, coverage for each chromosome, coverage for selected pathways and the coverage for genes of interest. Here, we focus on inflammatory and immunological pathways and genes relevant for haematopoietic stem cell transplantation. We compared three arrays: the Infinium Global Screening Array-24 v1.0, the Infinium OncoArray-500 K BeadChip and the Infinium PsychArray-24 v1.2 BeadChip. We employed the European population from the 1000 Genomes Project as reference genome. Global coverage was found to range between 12.2 and 14.2% whereas coverage for a selected pathway ranged from 6.2 to 13.2% and gene coverage ranged from 0 to 54.1%. The Global Screening Array outperformed both other arrays in terms of global coverage, for most chromosomes, most considered pathways and most genes. When selecting suitable arrays for a new study, the coverage of pathways or genes of interest should be considered in addition to global coverage. Local coverage should be regarded when discussing association findings inconsistent across studies and can be useful in data analysis and decision making for additional genotyping.
Similar content being viewed by others
Introduction
The importance of immune and inflammatory factors related to many common diseases is becoming more and more evident. Inflammation has long been associated with the development of cancer [1, 2]. Schmitt et al. [3] discovered that natural killer (NK) cells provide immune surveillance of cancer and Lanier [4] reported that their cytotoxic activity is controlled by a balance of activating and inhibitory signals.
Pahl [5] demonstrated the importance of NF-κB complexes in the activation by inflammatory cytokines and promotion of tumour-cell survival through anti-apoptotic signalling in prostate cancer [5, 6]. It is also known that Toll-like receptors play a key role in the innate immune system and have been found expressed by some types of tumour cells [7].
Previous genetic association studies have suggested that human leukocyte antigen (HLA) genes but also non-HLA genes may play an important role in haematopoietic stem cell transplantation (HSCT) and graft-versus-host disease (GvHD) [8]. HLA genes are encoded in a ~3500 kb segment on human chromosome 6p21.3, which is the most variable region in the human genome [8]. In addition, disturbances in immune and inflammatory-related systems have been associated with several psychiatric disorders [9].
Genome-wide association studies (GWAS) involving hundreds of thousands of genetic markers have led to the discovery of susceptibility genes for cancers and inflammatory diseases, as well as many other conditions and illnesses. As one of the first critical steps in developing such a large-scale study, researchers must choose the genotyping array to be implemented. As Delano et al. reported [10], the choice of genotyping platform is highly influential on the power of the study and thus on the likelihood of GWAS success. Thus, during the decision-making process, one needs to take into account a comparison of different single nucleotide polymorphism (SNP) arrays in consideration of the intended goal of the study. The most relevant and used criterion in SNP array evaluation is coverage. Global coverage is defined as the fraction of SNPs captured in terms of linkage disequilibrium (LD) by the SNPs on the array, representing the average level of coverage of all SNPs across the genome [11]. However, although global coverage provides us with an average evaluation of the array, it is not sufficient to capture variability in LD across the genome [12].
In order to achieve a fully informative estimate of coverage with respect to selected immune and inflammatory pathways and related genes, we evaluated three SNPs arrays from Illumina, Inc.: the Infinium Global Screening Array-24 v1.0 (GSA), the Infinium OncoArray-500 K BeadChip (OncoArray) and the Infinium PsychArray-24 v1.2 BeadChip (PsychArray). Our investigation provided estimates of global coverage across the genome, coverage for each chromosome, coverage for selected pathways and finally the coverage for genes of interest within these pathways. Pathways are selected based on their involvement in innate and adaptive immunity, cross talks with inflammation and haematopoietic stem cell transplantation. In order to follow this aim, we chose to investigate eight inflammatory and immunological pathways: natural-killer-cell-mediated cytotoxicity (hsa04650), NF-kappa B signalling (hsa04064), Wnt signalling (hsa04310), antigen processing and presentation (hsa04612), Toll-like receptor signalling (hsa04620), JAK-STAT signalling (hsa04630), insulin signalling (hsa04910) and B-cell receptor signalling (hsa04662). Europeans were used as reference sample in the analyses.
Materials and methods
The arrays
In order to compare arrays meaningfully, we selected only the arrays with at least 500 K markers. We estimated coverage for the GSA and two other consortia-derived customised arrays, the OncoArray and the PsychArray, all three manufactured by Illumina, Inc. These arrays are cost-effective, high-density arrays with at least 500 K SNPs and developed for large-scale genetic studies. Although our focus was on inflammatory and immune-related pathways, we did not consider the Infinium ImmunoArray-24 v2 BeadChip owing to its much smaller size of only 254 K SNPs. The arrays are summarised in Table 1.
Illumina developed the OncoArray in cooperation with cancer scientists forming the OncoArray Consortium. The array contains ~500 K SNPs, with 250 K proven tag SNPs covering the whole genome (backbone) and 250 K hand-selected SNPs of particular interest. It was built to provide insight into the relationship between gene variants predisposing to breast, ovarian, prostate, colorectal and lung cancer, the most relevant cancers in term of mortality [13, 14].
The PsychArray comprises 593 K markers and was developed in collaboration with the Psychiatric Genomics Consortium and several leading research institutions for genetic studies focussed on psychiatric predisposition and risk. The array contains ~271 K proven tag SNPs found on the Infinium Core-24 BeadChip, ~277 K markers from the Infinium Exome-24 BeadChip and ~50 K markers associated with common psychiatric disorders, such as schizophrenia, bipolar disorder, autism-spectrum disorders, attention deficit hyperactivity disorder, major depressive disorder, obsessive-compulsive disorder, anorexia nervosa and Tourette’s syndrome [15].
The GSA contains ~640 K SNPs and presents itself as a genomic tool for clinical research applications including disease risk profiling studies, pharmacogenomics research, wellness characterisation and complex disease discovery. The GSA has been optimised for unparalleled genomic coverage and imputation performance in all the five defined super populations (Africans, mixed Americans, East Asians, Europeans, South Asia) [16].
Only SNPs defined by rs-numbers were included in the analysis, since their genomic location is obtainable from the NCBI database, dbSNP [17]. Coverage will not be remarkable down biased using this filter, since the median MAF of the excluded markers e.g., for OncoArray is 0.03 in a sample of cancer-free Caucasian of the International Lung Cancer Consortium [18]. The three arrays have ~125 K SNPs in common. All information on the markers contained in the arrays is publicly available on the Illumina website [15, 16, 19].
The reference set
We estimated the coverage of these three Illumina arrays using the 1000 Genomes Project (Version 3 April 2012, NCBI Build 37) as reference set, as it is often adopted for imputation purposes. For our analysis we concentrated on the 286 European individuals (samples: GBR; IBS; CEU; TSI) and on SNPs with MAF ≥ 1%, calculating coverage based only on common SNPs. After MAF filtering, the final reference set included 8,846,061 SNPs on 23 chromosomes (1–22, X). In total, 13,725,914 SNP pairs were found to be in LD at a threshold of r2 ≥ 0.8.
The estimation equation
We calculated global coverage across the genome, global coverage for each chromosome, coverage for selected pathways and coverage for genes of interest. The coverage rate represents the fraction of all SNPs that can be captured by the array. We applied the equation:
defined by Barrett and Cardon [11] and Li et al. [12]. R is the number of SNPs in the reference set, T is the number of SNPs directly genotyped on the array and also given in the reference set, L is the number of SNPs in the reference set not on the array but in LD with a SNP on the array, r2 ≥ 0,8; G is the number of all SNPs validated in the dbSNP database with MAF ≥ 1%, 19 million and thus, 6809 SNPs in each 1 Mbp region. Since this equation ignores SNPs that are on the array but not in the reference set, we consider CR as the lower bound of coverage rate. We also computed a modified coverage estimate:
as proposed by Li et al. [12]; using their notation, we replace R1 = R1 + m, T1 = T + m, \({\mathrm{L}}_1 = \frac{{{\mathrm{T}}_1}}{{\mathrm{T}}}{\mathrm{L}}\), where m is the number of SNPs on the array not given in the reference set. Since a linear increase of tagged SNPs L with the number of SNPs on the array is implicitly assumed, CR1 tends to overestimate coverage [20]. Thus, we finally used the average of CR and CR1 as the final estimate.
The calculations were performed for the total sets of SNPs contained on each array and the sample of overlapping SNPs that the arrays have in common. The latter set of markers we defined as genome-wide, common non-customised backbone and is further denoted as the common backbone. Note that this common backbone has only approximately half of the sites of the Illumina backbone for OncoArray and PsychArray.
The reference set and the array data were reduced to the regions of interest in order to estimate coverage, except for global coverage. A gene was defined from the transcriptional start to end positions, including both exons and introns (with 50 Kbp upstream and downstream) [12]. Genes containing fewer than five SNPs were excluded from the analysis in order to prevent unreliable results [12, 20]. For the pathways, the reference and array data were reduced to the regions defined by all the genes in the pathway. The estimation was performed for pathways with more than five SNPs, in our case for any pathway of interest. All analyses were performed using the statistical computing language and environment R 3.4.1 [21].
The pathways and genes of interest
Since we had a special interest in HSCT, we primarily focussed on the following inflammatory and immunological pathways: natural-killer-cell-mediated cytotoxicity (hsa04650), NF-kappa B signalling (hsa04064), Wnt signalling (hsa04310), antigen processing and presentation (hsa04612), Toll-like receptor signalling (hsa04620), JAK-STAT signalling (hsa04630), insulin signalling (hsa04910) and B-cell-receptor signalling (hsa04662). The NF-κB signalling pathways have an essential role in many aspects of inflammation, innate and adaptive immunity. Importantly, NF-κB is also a crucial player in many steps of cell transformation. NF-κB cooperates with multiple other signalling molecules and pathways. Prominent nodes of crosstalk are mediated by Jak/STAT, WNT leading to mutual pathway activations or even negative regulations in a context dependent manner. The Jak/STAT pathway is also tightly linked to the regulation of inflammatory processes mediating the responses of immune cells to pro-inflammatory and anti-inflammatory cytokines. The capability of the host to build a defence against infections or danger signals key is related to the ability to processed and present antigens to immune cells and activates corresponding response cascades. It is well known that antigen presentation is mediated by MHC class I and class II molecules. These molecules are detectable on the surface of antigen presenting cells. The compatibility of these molecules is the key to the outcome of HSCT. Natural killer (NK) cells are large granular cells often lacking antigen specific cell surface receptors and involved in certain innate immune responses. In addition Toll-like receptors (TLRs) play crucial roles in the innate immune system mainly by activating different branches of NF-kB and IRF signalling. For those 37 genes that play a central role in these pathways, gene coverage was determined in addition. The Ensembl and KEGG databases were consulted to annotate SNPs to genes and genes to pathways, respectively [22, 23]. The positions of the markers on the arrays and annotation files correspond to Genome Reference Consortium Human Build 37 (GRCh37).
Results
Details of global coverage and coverage for each chromosome across the arrays studied are depicted in Table 2. The global coverage rates were estimated as 14.23% for the GSA, 12.51% for the OncoArray and 12.24% for the PsychArray. The global coverage estimated for the OncoArray and PsychArray is comparable with the findings of Ha et al. [20] for arrays similar in size, considering the same genetic super population. GSA was the best most of the time. The range of variation for coverage between chromosomes is 10.44–13.52% for GSA, 9.63–13.93% for the OncoArray, and 9.92–12.56% for the PsychArray. Chromosomes 4, 5, 7 and 14 consistently have coverage below 11% for all of the three arrays studied. Note also that the coverage of chromosome 19 for the PsychArray is lower than 10%. Nevertheless, there are two genes on chromosome 19, HCST and TYROBP, particularly well covered by all three arrays (24.32 and 21.02% for GSA, 43.77% and 40.83% for OncoArray, and 25.21 and 21.72% for PsychArray; these two do not belong to the pathways of interest).
Table 3 illustrates the details on coverage for pathway and number of SNPs for each array. The coverage for the eight pathways of interest ranges from 6.21% (PsychArray for hsa04612) to 13.91% (GSA for hsa04064). With one exception, GSA always outperformed the competing arrays, while the differences between the arrays remain relatively small. Specifically, the range for coverage between the considered pathways is 8.11–13.91% for GSA, 8.65–12.04% for OncoArray and 6.21–11.35% for PsychArray. However, across the eight pathways of interest, there are 16 genes for GSA, 22 for OncoArray and 24 for PsychArray with coverage of less than 1%. The estimates of pathway coverage differ little between the two equations CR and CR1, in median 1.44 percentage points. The maximum difference was observed for the coverage of hsa04612 by the OncoArray, with CR = 4.2% and CR1 = 13.1% (see Supplementary Material).
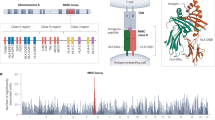
The distribution of the gene coverage within each pathway and for each array is displayed in Fig. 1. In comparison, GSA performs the best on average. However, a strong variability in gene coverage can be easily recognised across all arrays. Individual genes reach coverage of more than 50% in all but two pathways (hsa04612/hsa04620); it is worth noting that the best results were obtained with the OncoArray. Most prominent is pathway hsa04612, which contains four genes of the HLA group well covered by GSA and the OncoArray: HLA-DQA1 (6: 32595956-32614839) with 54.08% (GSA) and 19.42% (OncoArray), HLA-DQB1 (6: 32627244-32636160) with 51.49% and 24.18%, also HLA-C (6: 31236526-31239907) with 23.85% and 40.05% and HLA-DOB (6: 32780540-32784825) with 29.49 and 38.45%.

Gene coverage by array within selected pathways. Distribution of gene coverage for the following pathways: hsa04650: natural-killer-cell-mediated cytotoxicity, hsa04064 NF-kappa B signalling, hsa04310 Wnt signalling, hsa04612 antigen processing and presentation, Toll-like hsa04620 receptor signalling, hsa04630 JAK-STAT signalling, hsa04910 insulin signalling, hsa04662 B-cell-receptor signalling. GSA Infinium Global Screening Array-24 v1.0, OncoArray Infinium OncoArray-500 K BeadChip, PsychArray InfiniumPsychArray-24 v1.2 BeadChip
Details of coverage for the 37 genes of interest are presented in Table 4. The coverage for the 37 genes of interest (see Table 4) has a range from 0.28% (OncoArray for CCR5) to 39.48% (OncoArray for IL10RA). The greatest variation in coverage was found for the gene MICA (GSA: 27.34%, OncoArray: 25.10%, PsychArray: 7.82%), the least for TNFSF13 (GSA: 7.78%, OncoArray: 8.11%, PsychArray: 7.06%). For most genes, GSA proved to have the highest coverage; in only five out of 37 genes was GSA the worst of the three arrays studied in terms of coverage. In general, the estimates of gene coverage differ little between the two equations CR and CR1, in median 1.18 percentage points. However, a maximum difference was observed for the coverage of MICA by the GSA, with CR = 9.2% and CR1 = 45.5%. Similar strong deviations were also observed for LTA, LTB, MICB and TNF. All these relatively short genes are located at 6p21.33 within the human major histocompatibility complex (MHC), known for its long LD structure [24] (see Supplementary Material).
Lastly, the common backbone (125 K SNPs overlapping between the arrays) accounts for 5.47% global coverage. This compares to 12.24–14.23% global coverage using all SNPs of an array, thus this SNP set common to all arrays is clearly capturing not even half of the global coverage of the arrays. Coverage across chromosomes in the common backbone ranges from 4.26% to 5.82% (see Table 2). However, the common backbone accounts for no more than 0.04% of coverage (hsa 04310) across pathways (see Table 3). Nevertheless, the gene coverage of the common backbone ranges between 0% (CD40LG) and 20.93% (IL10RA) (see Table 4).
Discussion
We calculated the global coverage for three high-throughput genotyping arrays manufactured by Illumina, Inc., as well as the coverage by chromosome, the gene coverage and coverage for eight selected inflammatory and immune pathways. Analogous to global coverage, gene coverage is the fraction of SNPs on the array and allocated in a gene region, representing the average level of coverage of all SNPs of 1000 Genomes reference set in the same region (with fraction captured in terms of LD). The same applies to chromosomes and to pathways, where the “region” may be scattered across the genome.
The three arrays demonstrated noteworthy variations in their coverage, not only in general but also with respect to inflammatory and immune response genes and pathways.
Our estimates of coverage indicate better performance of the GSA compared to the OncoArray and the PsychArray globally, for most of the chromosomes and for most considered inflammatory and immunological pathways and genes. However, it has to be stated that the improvement in global coverage of about 2% of the GSA may result from the fact that it is larger in size. Nevertheless, this improvement in coverage is small, considering that the GSA contains twice as many rs-SNPs (~627 K) than the OncoArray (~343 K) or PsychArray (~306 K) and the loss by filtering for rs-numbers was lowest for the GSA.
We believe that researchers interested in specific regions of the genome will be able to use our approach to choose the array that best fits their goals. If interested in specific molecular mechanisms or gene families, the emphasis should be placed on pathway or gene coverage rather than on SNP numbers and global coverage. For relatively short genes in regions of low or long-range LD, it seems advisable to examine the lower and upper limits of coverage in addition to their mean.
Low coverage of a gene may result in low power to detect a genetic association, which can often lead to the wrong scientific conclusion that there is no association. Another aspect related to coverage is the possibility of inconsistency in the results between comparable GWAS, up to heterogeneous marker selection for polygenic risk scores [12]. Considering coverage can also be informative for identify markers not included in an already used array but worth to be extra genotyped by e.g., PCR-based methods. One can also exclude genes or pathways of low-coverage from the analysis of already genotyped samples to reduce the burden of multiple tests. Even though it appears that the GSA generally performs best, no general recommendation can be made because of the sample used for comparison. We focussed on a selection of inflammatory and immune pathways. We considered only genotyping of Caucasians with only one reference sample. The extension to other molecular mechanisms, other populations and other reference samples may well affect the results. We therefore recommend calculating global coverage, pathway and gene coverage for the focused pathways and genes of the planned study, but at least for the envisaged population.
To conclude, global coverage alone does not provide enough information to choose the most appropriate genotyping array for a study in planning. Pathway or gene coverage should be considered instead. Moreover, local coverage needs to be regarded when discussing inconsistencies in the findings between GWAS and can be useful in data analysis and decision making for additional genotyping.
References
Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow? Lancet Lond Engl. 2001;357:539–45.
Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–7.
Schmitt C, Ghazi B, Bensussan A. NK cells and surveillance in humans. Reprod Biomed Online. 2008;16:192–201.
Lanier LL. NK cell recognition. Annu Rev Immunol. 2005;23:225–74.
Pahl HL. Activators and target genes of Rel/NF-kappaB transcription factors. Oncogene. 1999;18:6853–66.
Murphy KM, Weaver C. Janeway immunologie. 9th edn. Springer Spektrum, Berlin; 2008. Available from: https://www.springer.com/de/book/9783662560037. Accessed 19 Oct 2018.
Droemann D, Albrecht D, Gerdes J, Ulmer AJ, Branscheid D, Vollmer E, et al. Human lung cancer cells express functionally active Toll-like receptor 9. Respir Res. 2005;6:1.
Shiina T, Hosomichi K, Inoko H, Kulski JK. The HLA genomic loci map: expression, interaction, diversity and disease. J Hum Genet. 2009;54:15–39.
Leboyer M, Berk M, Yolken RH, Tamouza R, Kupfer D, Groc L. Immuno-psychiatry: an agenda for clinical practice and innovative research. BMC Med. 2016;14:173.
Delano D, Eberle M, Galver L, Rosenow C. Array differences in genomic coverage and data quality impact on GWAS success. https://www.illumina.com/events/webinar/archive/web_Nov192008_183.html. Accessed 18 Oct 2018.
Barrett JC, Cardon LR. Evaluating coverage of genome-wide association studies. Nat Genet. 2006;38:659–62.
Li M, Li C, Guan W. Evaluation of coverage variation of SNP chips for genome-wide association studies. Eur J Hum Genet Ejhg. 2008;16:635–43.
Amos CI, Dennis J, Wang Z, Byun J, Schumacher FR, Gayther SA, et al. The OncoArray Consortium: a network for understanding the genetic architecture of common cancers. Cancer Epidemol Prev Biomark. 2017;26:126–35.
Infinium OncoArray-500K BeadChip. Data Sheet: Cancer Genomic [Internet]. [cited 2018 Oct 18]. Available from: https://www.illumina.com/content/dam/illumina-marketing/documents/products/datasheets/datasheet_oncoarray500k.pdf
Infinium PsychArray-24 Kit. Psychiatric predisposition microarray. https://www.illumina.com/products/by-type/microarray-kits/infinium-psycharray.html. Accessed 18 Oct 2018.
Infinium Global Screening Array-24 Kit. Population-scale genetics. https://www.illumina.com/products/by-type/microarray-kits/infinium-global-screening.html. Accessed 18 Oct 2018.
Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, et al. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29:308–11.
McKay JD, Hung RJ, Han Y, Zong X, Carreras-Torres R, Christiani DC, et al. Large-scale association analysis identifies new lung cancer susceptibility loci and heterogeneity in genetic susceptibility across histological subtypes. Nat Genet. 2017;49:1126–32.
Infinium OncoArray-500K BeadChip. Research cancer predisposition and risk. https://www.illumina.com/products/by-type/microarray-kits/infinium-oncoarray-500k.html. Accessed 18 Oct 2018.
Ha N-T, Freytag S, Bickeböller H. Coverage and efficiency in current SNP chips. Eur J Hum Genet. 2014 Sep;22:1124–30.
R: The R project for statistical computing. https://www.r-project.org/. Accessed 22 Oct 2018.
KEGG PATHWAY database. https://www.genome.jp/kegg/pathway.html. Accessed 18 Oct 2018.
Cunningham F, Amode MR, Barrell D, Beal K, Billis K, Brent S, et al. Ensembl 2015. Nucleic Acids Res. 2015;43(Database issue):D662–669.
Ahmad T, Neville M, Marshall SE, et al. Haplotype-specific linkage disequilibrium patterns define the genetic topography of the human MHC. Hum Mol Genet. 2003;12:647–56.
Acknowledgements
This project was conducted as collaboration with the Transdisciplinary Research of Cancer in Lung and the International Lung Cancer Consortium (TRICL/ILCCO). We also wish to thank Andrew Entwistle for proofreading the paper.
Funding
The National Institutes of Health (7U19CA203654-02/114564-5111078 Integrative Analysis of Lung Cancer Etiology and Risk) supported this work.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Tozzi, V., Rosenberger, A., Kube, D. et al. Global, pathway and gene coverage of three Illumina arrays with respect to inflammatory and immune-related pathways. Eur J Hum Genet 27, 1716–1723 (2019). https://doi.org/10.1038/s41431-019-0441-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-019-0441-2
This article is cited by
-
Iam hiQ—a novel pair of accuracy indices for imputed genotypes
BMC Bioinformatics (2022)
