
Abstract
Colorectal cancer (CRC) is one of the major cancer types in the western world including Sweden. However, known genetic risk factors could only explain a limited part of heritability of the disease. Moreover, colon and rectal cancers are habitually discussed as one entity, colorectal cancer, although different carcinogenesis has been recognized. A genome-wide linkage scan in 32 colon- and 56 rectal cancer families from Sweden was performed based on 475 non-FAP/HNPCC patients genotyped using SNP arrays. A maximum HLOD of 2.50 at locus 6p21.1-p12.1 and a HLOD of 2.56 at 18p11.2 was obtained for colon and rectal cancer families, respectively. Exome sequencing over the regions of interest in 12 patients from six families identified 22 and 25 candidate risk variants for colon and rectal cancer, respectively. Haplotype association analysis in the two regions was carried out between additional 477 familial CRC cases and 4780 controls and suggested candidate haplotypes possibly associated with CRC risk. This study suggested two new linkage regions for colon cancer and rectal cancer with candidate predisposing variants. Further studies are required to elucidate the pathogenic mechanism of these regions and to pinpoint the causative genes.
Similar content being viewed by others


Introduction
More than 1.2 million new cases are diagnosed with colorectal cancer (CRC) in the world yearly, primarily in the western world [1]. In Sweden, CRC is the third most common cancer type among women and men, and affects 4000–6000 individuals each year [2]. Less than 5% of CRC cases are caused by known genes, such as those causing familial adenomatous polyposis (FAP) and hereditary non-polyposis colorectal cancer (HNPCC) [3]. Previous CRC genes mapped using linkage analysis include APC [4], MSH2 [5], and MLH1 [6]. Other linkage studies have suggested potential CRC loci at 2q, 3q, 4q, 8q, 9q, 10p, 12q, 14q, and 15q [7,8,9,10,11,12,13,14]. Moreover, hundreds of common variants have been reported by genome-wide association studies (GWAS) to be associated to CRC, but they only describe a limited portion of disease risk [15, 16]. Altogether, germline variants in known genes and moderate- and low risk variants were suggested to explain 10–15% of the genetic CRC contribution [17]. Although parts of the causative CRC genetic factors are known, further investigation to learn about the missing genetics is important, since up to 35% of all CRC cases could be explained by hereditable factors [18].
Colon cancer and rectal cancer are habitually discussed as colorectal cancer. The question whether it is one single or two different entities has been under debate. Some studies have presented possible differences and recognized that colon and rectal cancer have different carcinogenesis. Bufill et al. reported that the location of the tumor might be a marker for the clinical feature [19]. Tumors arise predominantly distal to the splenic flexure in adenomatous polyposis, while in HNPCC, most tumors arise proximal to the splenic flexure [19]. One study disclosed a higher frequency in genetic alteration and allelic losses on chromosome 5q, 17p and 18 among distal compared to proximal colorectal tumors [20]. Kapiteijn et al. reported that rectal cancers had a significantly higher immunohistochemical expression of TP53 and nuclear β-catenin compared to colon cancers and that TP53 mutation rate was higher in rectal cancer cases [21]. However, no significant differences were seen for clinical and histopathological data [21]. Another study showed that KRAS variants in stool DNA were associated with tumors in the sigmoid colon and rectum but not with tumors from other parts of the colon [22]. Elevated expression of the oncogene MYC was seen more often in the left-side (rectum, sigmoid and descending colon) compared to the right-side (caecum and ascending colon) of colorectum [23].
Therefore, novel loci harboring predisposing genes could possibly be found by analyzing colon and rectal cancer families separately. Thus, we performed a new linkage scan on 32 colon cancer and 56 rectal cancer families corresponding to 169 and 306 individuals, respectively. These families were included in a genome-wide linkage analysis of 121 families conducted previously [9]. In order to find evidence supporting the candidate regions revealed by linkage analysis and to further pinpoint genes and variants that potentially affect functions, we performed targeted exome sequencing over the two regions in the families mostly contributing to the positive LOD scores and haplotype association analysis in additional CRC cases and controls.
Materials and methods
Study subjects
Cancer families were recruited through the Department of Clinical Genetics at the Karolinska University Hospital in Stockholm, Sweden between 1990 and 2005. FAP was excluded using medical records from affected individuals and HNPCC was excluded using our current clinical protocols [24]. Families were included in the study if there were at least two affected relatives informative for linkage analysis (i.e., at least a sib-pair). A family was included in the linkage analysis if the family could be classified to have a risk for colon or rectal cancer. Eighty-eight of the previously analyzed 121 families fulfilled the criteria above and were included in the linkage analysis (Table 1).
A case–control study used 477 familial CRC cases from the Swedish Colorectal Cancer Low-risk Study and 4780 control individuals from the Swedish Twin Registry [25, 26]. The 477 CRC cases were from a cohort of more than 3300 consecutive patients operated on for CRC in 14 hospitals in and around Stockholm and Uppsala between 2004 and 2009. For the twin controls, phenotypic data on cancer had previously been obtained through linking the twins to the Swedish Cancer Registry using the unique person identification number available for all Swedish citizens. Only one twin from each twin pair where none was affected was considered eligible for serving as control in the association analysis.
The study was undertaken in agreement with the Swedish legislation of ethical permission (2003:460) and according to the decision in the Stockholm regional ethical committee (2008/125-31.2 and 02-489). All participants had given informed consent to participate in the study.
Genotyping and quality control (QC)
Genomic DNA was extracted from peripheral blood using standard procedures. Genotyping was performed as previously described [9].
In order to generate haplotypes for CRC families, a total of 60 parent–child pairs from 10 colon cancer families, 17 rectal cancer families as well as 33 CRC families without a clear tumor location predominance from this study was genotyped with the Illumina Infinium HumanOmniExpress-12v1 BeadChip (730,525 markers) at the SNP&SEQ Technology Platform in Uppsala, Sweden. The overall reproducibility of the genotype data was 99.996% based on 1.53% of duplicated genotyping, with an average call rate per SNP of 99.43%.
The 477 additional familial CRC cases were genotyped using the Illumina Infinium OncoArray-500K BeadChip at the Center for Inherited Disease Research at Johns Hopkins University, MD, US [16]. The 4780 controls from the Swedish TwinGene registry were genotyped using the Illumina OmniExpress BeadChip in Uppsala, Sweden. All samples went through initial QC at their corresponding centers before being merged on the 235,573 SNPs that were shared between the two platforms. QC of the merged dataset excluded variants from analysis if call rate was ≤97%, minor allele frequency was <1% or if the variant deviated significantly from Hardy–Weinberg equilibrium (p ≤ 1E−7). Samples were removed in case of genotyping success rate was <97%, gender discrepancy between reported and X-chromosome heterozygosity-predicted, abnormal heterozygosity (>3 standard deviations from mean) or detection of cryptic relatedness.
Linkage analysis
Pedcheck [27] was used to check for the initial Mendelian inheritance analysis among the families. The family-based genetic model was used for parametric linkage analysis for all chromosomes including chromosome X. Non-parametric analysis was performed as a supplement. LOD scores as well as heterogeneity LOD scores were computed using MERLIN (version 1.1.2) [28] and was given for all genotyped positions. Analyses were done assuming both dominant and recessive traits and the parameters were set as described by our previously published paper [9]. Individuals with CRC or a polyp with high degree dysplasia were coded as affected. All subjects in the 88 families from the two genotyping sessions were included in the analysis. Due to two genotyping sets, two map files were merged and 7256 markers were used in the analysis. As a consequence of limitations in MERLIN, four large families had to be split when running the analysis.
Exome sequencing and variant calling
Twelve patients from six families, three colon (110, 301, 350) and three rectal (8, 918, 1213) cancer families, respectively, were selected for exome sequencing based on their major contribution to the LOD scores in the linkage regions. In four families two affected sibs were sequenced. In one family a single patient was sequenced and in the last family three sibs were subject to sequencing.
Sequencing libraries were prepared from genomic DNA using TruSeq DNA Sample Preparation Kit (Illumina, San Diego, CA, USA) or SureSelectXT Reagent HSQ 96 Auto kit (Agilent, Santa Clara, CA, USA) according to manufacturers’ instructions. Exome enrichment was performed using TruSeq Exome Enrichment Kit (Illumina) or SureSelect XT Human All Exon V5 library (Agilent). Multiplexed paired-end libraries were pooled in equal molar and sequenced on an Illumina HiSeq 2000 or HiSeq 2500 system (Illumina) according to manufacturer’s instructions.
Base calling was performed on the instrument with RTA (1.12.4.2 or 1.13.48) and the resulting BCL files were filtered, de-multiplexed, and converted to FASTQ format using CASAVA 1.7 or 1.8 (Illumina). Raw reads were mapped to the hg19 GRCh37 reference genome sequence using bwa (0.5.9), and variants were called using GATK (1.0.5974) following realignment and recalibration. Variant annotation was performed using ANNOVAR (released 2013-08-23).
Haplotype association analysis
Association analysis were carried out between 477 familial CRC cases and 4780 controls over the two regions of interest revealed by linkage analysis in sliding windows containing 1–25 consecutive markers. In short, haplotype frequency was estimated for each window and p-values were calculated using Plink v.1.07 [29].
Data deposition
Non-synonymous coding sequence variants with a MAF < 0.20 that segregated in at least one of the six selected families with corresponding disease information were deposited to the gene variant database of Leiden Open Variation Database (https://databases.lovd.nl/shared/genes). Individual IDs were #00208599, #00208600, #00208601, #00208603, #00208611, and #00208612 for one representative from each of the families 310, 110, 350, 8, 918, and 1213, respectively.
Results
Linkage analysis suggested candidate regions for colon and rectal cancer separately
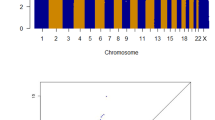
A total of 88 families were genotyped and analyzed in two groups, comprising of 32 colon and 56 rectal cancer families with 306 and 169 individuals, respectively (Table 1). No LOD or HLOD score above three was observed. However, suggestive linkage could be demonstrated for colon as well as rectal cancer families (Fig. 1). Regions with HLODs above 1.0 are summarized in Table 2. A maximum HLOD of 2.5 was observed for a 6 Mb region at locus 6p21.1-p12.1 in the colon cancer families. The highest HLOD was 2.6 for the rectal cancer families at locus 18p11.2 with about 10 Mb in length.

LOD/HLOD score plots for colon and rectal cancer families. a LOD/HLOD plot for 32 colon cancer families. b LOD/HLOD plot for 56 rectal cancer families. LODs are represented in red and HLODs are represented in cyan
Exome sequencing revealed genetic variants segregating in cancer families
In order to identify variants that possibly affect gene function in the linked regions, we did exome sequencing on twelve individuals representing the families contributing to the LOD scores. Three colon cancer families were included to investigate the candidate region on chromosome 6p21.1, whereas three rectal cancer families were included for the region on chromosome 18p11.2. Non-synonymous coding variants with a MAF < 0.20 in the regions of interest were assessed in relevant family members. We report variants segregating in all individuals for at least one of the three families (Tables 3 and 4). Twenty-two variants in 18 genes and 25 variants in 10 genes were identified segregating in colon cancer and rectal cancer families, respectively. Among the 22 variants observed in the colon cancer patients, 20 were missense variants, one was a frameshift insertion and one was an in-frame deletion. The 25 variants in the rectal cancer families were all missense variants.
Haplotype association analysis identified candidate targets
To further pinpoint the genetic risk factors for colon and rectal cancers, we performed haplotype association analysis on the two regions of suggestive linkage (HLOD > 2). A total of 593 and 554 SNPs was successfully genotyped in the two regions on chromosomes 6 and 18, respectively. Association analysis between 477 familial CRC cases and 4780 controls using these genetic markers identified two candidate risk loci on chromosome 6 and two on chromosome 18. At least one candidate risk haplotype of each loci was associated with an elevated CRC risk (odds ratio 1.68–2.45) with a p-value lower than 1E−4. One of these four candidate risk haplotypes was relatively common (haplotype frequency of 15% in the control group), whereas the other three were infrequent (haplotype frequency 2–5% in the control group) (Fig. 2).

Candidate risk haplotypes revealed by sliding-window association analysis within the linked regions on chromosome 6 (a) and chromosome 18 (b). Association was evaluated for haplotypes of sizes ranging from 1 to 25 markers between 477 familial CRC cases and 4 780 controls. All haplotypes with OR > 1 and p-value < 1E−4 were listed with p-value, odds ratio (OR), estimated frequency in controls (F_U) and cases (F_A). One haplotype of highest interest (lowest p-value and highest OR) for each of the four loci was indicated in orange and searched among 60 CRC families. Familial haplotypes of the most informative families potentially carrying these haplotypes were listed (question marks indicate undetermined markers of the haplotypes). Genomic regions covered by these risk haplotypes were illustrated showing co-localized genes, where exons and introns were indicated with dark and light gray, respectively
One risk haplotype identified on chromosome 6 stretched 14 kb in size, contained 4 markers and overlapped with gene KCNK5 (Fig. 2a). Haplotyping of 60 CRC families revealed at least one colon cancer family (family 110), one rectal cancer family (family 242) and one CRC family without tumor site predominance (family 869) that potentially have this haplotype (Fig. 2a). It is notable that two of the three colon cancer families that contributed to the LOD score, families 110 and 301 (data not shown), were identified as potential carriers of this haplotype. The other risk haplotype on chromosome 6 was 176 kb in size and overlapped with genes CDC5L, SPATS1 and part of TMEM151B. At least two colon cancer families (families 46 and 237) likely harbor this haplotype (Fig. 2a).
One risk haplotype on chromosome 18 was 61 kb in size but did not overlap with any known gene. Two rectal cancer families (families 425 and 1252) and one colon cancer family (family 68) in our study clearly harbored this haplotype (Fig. 2b). One of the most linked rectal cancer families, family 918, is also a potential carrier of this haplotype (data not shown). The other risk haplotype on chromosome 18 overlapped with part of the gene PIEZO2, and at least one rectal cancer family (family 1425) is likely to have this 220-kb haplotype based on genotyping of the parent–child pair (Fig. 2b).
Discussion
CRC is a multifactorial disease. Previous studies have shown that tumor location differs among FAP compared to HNPCC patients and that different tumor sites would display diverse genetic alterations and allelic loss at 5q, 17p and 18 [19, 20]. Also, gene expressions and mutation rates vary among right and left colon and rectal tumors [21,22,23]. We hypothesized that, by subdividing the CRC families into colon and rectal cancer families, it would hopefully result in novel loci and predisposing genes for the two different cancer entities.
Our linkage analysis provided us with some interesting regions with suggestive linkage HLOD = 2.5 for the colon cancer families on locus 6p21.1-p12.1 and HLOD = 2.6 for the rectal cancer families on locus 18p11.2 (Table 2). These regions have not yet, to our knowledge, been reported by other linkage studies, possibly because no previous study subdivided the CRC families into colon cancer and rectal cancer families.
Exome sequencing was carried out on twelve individuals representing the families contributing to the LOD scores and identified within the linked regions 22 colon and 25 rectal cancer variants segregating in the cancer families, respectively (Tables 3, 4). Genes harboring these variants are involved in signal transduction (GLP1R, CCND3, CUL7, PTK7, VAPA, APCDD1, MC5R, ROCK1, NPC1), microtubule-based process (DNAH8, KIF6, CUL9, CEP192), RNA metabolism (APOBEC2, USP49, BYSL, XPO5), establishment of localization (PEX6, GTPBP2 and PIEZO2) and cell differentiation (LRFN2, MEA1, LAMA3) among others. Some of these genes have been implicated in colorectal tumorigenesis, for instance, CCND3 is a known oncogene in multiple cancer types including CRC [30]. PTK7, whose variants presented in two families, is reported to be expressed and actively involved in various malignancies including CRC, and its function in the Wnt signaling pathway has been demonstrated (reviewed in the ref. [31]). Moreover, overexpression of PTK7 has been implicated as a biomarker for adenoma and CRC, and is correlated with several clinicopathological features such as TNM stage, tumor differentiation, lymph node and distant metastasis [32, 33]. Similarly, GTPBP2 is also known as a positive regulator of the Wnt signaling pathway [34], which is involved in tumorigenesis of a wide variety of cancers including CRC. The variant in the gene APCDD1 is also shared among two families. APCDD1 is suggested to be regulated by the β-catenin/Tcf complex involved in colorectal tumorigenesis [35]. A previous methylation microarray-based scanning has revealed that hypermethylation of GLP1R is a biomarker for CRC and adenoma [36]. CUL7 has been identified as an oncogene, since it could directly bind to p53 and prevent cells from Myc-induced apoptosis [37]. Overexpression of CUL7 could distinguish metastatic CRC samples from the non-metastatic ones [38]. XPO5 is a key protein responsible for miRNA transportation and is upregulated both at mRNA and protein levels in CRC. Its overexpression is associated with worse clinicopathologic features and poor survival in CRC [39]. The POTEC gene had one variant shared in two families and other variants in single families. POTEC is a member of the highly homologous POTE family which are expressed in multiple cancer types including colon cancer [40, 41]. Gene ROCK1 is part of the Rho-kinase family and is overexpressed in CRC cell lines [42] and tissues [43]. Overexpression of ROCK1 has been shown to lead to increased CRC cell proliferation, transformation and invasion [42]. The gene CTAGE1 is described as a cancer antigen for T-cell lymphoma and other malignancies [44], and is expressed in 12–19% CRCs [45]. Previous studies have reported somatic frameshift variants of LAMA3 in CRC with high microsatellite instability [46] and deletions of the LAMA3 gene in CRC with high chromosomal instability [47].
Haplotype analysis has been proven valuable in identifying susceptibility genes in familial breast cancer [48] and cancer syndromes [49], especially in populations with a relatively homogenous genetic background. In particular, a candidate CRC locus on chromosome 9q [8, 9, 13, 14] was recently suggested to be explained by two different risk haplotypes in familial and sporadic bowel cancer [50]. In order to search for additional support of the two loci in the current study and to further pinpoint candidate risk variants, we performed haplotype association studies between familial CRC cases and controls for the two regions. The four candidate haplotypes harbor coding regions of several genes including CDC5L (cell division cycle 5 like), a positive regulator of cell cycle G2/M progression and key promoter of colorectal cancer cells [51]. The relationship between colorectal cancer and other genes located within these candidate haplotypes haven’t been well studied. But the fact that some of the families in the linkage analysis were demonstrated to be potential carriers of these risk haplotypes supports that these haplotypes may by associated with an increased risk.
In conclusion, we propose two new linkage regions for colon cancer and rectal cancer. Haplotype analysis provides additional support and information regarding candidate variants that might affect function. We also report candidate variants within the linked regions that possibly predispose to CRC risk. Further studies on these genes of interest are needed to support or exclude them to be harboring disease causing variants.
References
Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. Cancer J Clin. 2011;61:69–90.
Socialstyrelsen. Cancer incidence in Sweden 2011=Cancerförekomst i Sverige 2011. Stockholm: Socialstyrelsen; 2012.
Lynch HT, Lynch PM, Lanspa SJ, Snyder CL, Lynch JF, Boland CR. Review of the Lynch syndrome: history, molecular genetics, screening, differential diagnosis, and medicolegal ramifications. Clin Genet. 2009;76:1–18.
Bodmer WF, Bailey CJ, Bodmer J, Bussey HJ, Ellis A, Gorman P, et al. Localization of the gene for familial adenomatous polyposis on chromosome 5. Nature. 1987;328:614–6.
Peltomaki P, Aaltonen LA, Sistonen P, Pylkkanen L, Mecklin JP, Jarvinen H, et al. Genetic mapping of a locus predisposing to human colorectal cancer. Science. 1993;260:810–2.
Lindblom A, Tannergard P, Werelius B, Nordenskjold M. Genetic mapping of a second locus predisposing to hereditary non-polyposis colon cancer. Nat Genet. 1993;5:279–82.
Cicek MS, Cunningham JM, Fridley BL, Serie DJ, Bamlet WR, Diergaarde B, et al. Colorectal cancer linkage on chromosomes 4q21, 8q13, 12q24, and 15q22. PLoS One. 2012;7:e38175.
Kemp ZE, Carvajal-Carmona LG, Barclay E, Gorman M, Martin L, Wood W, et al. Evidence of linkage to chromosome 9q22.33 in colorectal cancer kindreds from the United Kingdom. Cancer Res. 2006;66:5003–6.
Kontham V, von Holst S, Lindblom A. Linkage analysis in familial non-lynch syndrome colorectal cancer families from sweden. PLoS One. 2013;8:e83936.
Picelli S, Vandrovcova J, Jones S, Djureinovic T, Skoglund J, Zhou XL, et al. Genome-wide linkage scan for colorectal cancer susceptibility genes supports linkage to chromosome 3q. BMC Cancer. 2008;8:87.
Roberts A, Nancarrow D, Clendenning M, Buchanan DD, Jenkins MA, Duggan D, et al. Linkage to chromosome 2q32.2-q33.3 in familial serrated neoplasia (Jass syndrome). Fam Cancer. 2011;10:245–54.
Saunders IW, Ross J, Macrae F, Young GP, Blanco I, Brohede J, et al. Evidence of linkage to chromosomes 10p15.3-p15.1, 14q24.3-q31.1 and 9q33.3-q34.3 in non-syndromic colorectal cancer families. Eur J Hum Genet. 2012;20:91–6.
Skoglund J, Djureinovic T, Zhou XL, Vandrovcova J, Renkonen E, Iselius L, et al. Linkage analysis in a large Swedish family supports the presence of a susceptibility locus for adenoma and colorectal cancer on chromosome 9q22.32-31.1. J Med Genet. 2006;43:e7.
Wiesner GL, Daley D, Lewis S, Ticknor C, Platzer P, Lutterbaugh J, et al. A subset of familial colorectal neoplasia kindreds linked to chromosome 9q22.2-31.2. Proc Natl Acad Sci USA. 2003;100:12961–5.
Cheng TH, Thompson D, Painter J, O’Mara T, Gorman M, Martin L, et al. Meta-analysis of genome-wide association studies identifies common susceptibility polymorphisms for colorectal and endometrial cancer near SH2B3 and TSHZ1. Sci Rep. 2015;5:17369.
Schmit SL, Edlund CK, Schumacher FR, Gong J, Harrison TA, Huyghe JR, et al. Novel common genetic susceptibility loci for colorectal cancer. J Natl Cancer Inst. 2019;111(2):146-157.
Whiffin N, Houlston RS. Architecture of inherited susceptibility to colorectal cancer: a voyage of discovery. Genes. 2014;5:270–84.
Lichtenstein P, Holm NV, Verkasalo PK, Iliadou A, Kaprio J, Koskenvuo M, et al. Environmental and heritable factors in the causation of cancer—analyses of cohorts of twins from Sweden, Denmark, and Finland. N Engl J Med. 2000;343:78–85.
Bufill JA. Colorectal cancer: evidence for distinct genetic categories based on proximal or distal tumor location. Ann Intern Med. 1990;113:779–88.
Delattre O, Olschwang S, Law DJ, Melot T, Remvikos Y, Salmon RJ, et al. Multiple genetic alterations in distal and proximal colorectal cancer. Lancet. 1989;2:353–6.
Kapiteijn E, Liefers GJ, Los LC, Kranenbarg EK, Hermans J, Tollenaar RA, et al. Mechanisms of oncogenesis in colon versus rectal cancer. J Pathol. 2001;195:171–8.
Frattini M, Balestra D, Pilotti S, Bertario L, Pierotti MA. Tumor location and detection of k-ras mutations in stool from colorectal cancer patients. J Natl Cancer Inst. 2003;95:72–3. author reply 3
Rothberg PG, Spandorfer JM, Erisman MD, Staroscik RN, Sears HF, Petersen RO, et al. Evidence that C-Myc expression defines 2 genetically distinct forms of colorectal adenocarcinoma. Br J Cancer. 1985;52:629–32.
Lagerstedt Robinson K, Liu T, Vandrovcova J, Halvarsson B, Clendenning M, Frebourg T, et al. Lynch syndrome (hereditary nonpolyposis colorectal cancer) diagnostics. J Natl Cancer Inst. 2007;99:291–9.
Forsberg A, Keranen A, VONH S, Picelli S, Papadogiannakis N, Ghazi S, et al. Defining new colorectal cancer syndromes in a population-based cohort of the disease. Anticancer Res. 2017;37:1831–5.
Magnusson PK, Almqvist C, Rahman I, Ganna A, Viktorin A, Walum H, et al. The Swedish Twin Registry: establishment of a biobank and other recent developments. Twin Res Hum Genet. 2013;16:317–29.
O’Connell JR, Weeks DE. PedCheck: a program for identification of genotype incompatibilities in linkage analysis. Am J Hum Genet. 1998;63:259–66.
Abecasis GR, Cherny SS, Cookson WO, Cardon LR. Merlin-rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet. 2002;30:97–101.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–75.
Watson DS, Brotherick I, Shenton BK, Wilson RG, Angus B, Varma JS, et al. Cyclin D3 expression, cell proliferation and pathological stage of human primary colorectal cancer. Oncology. 1999;56:66–72.
Dunn NR, Tolwinski NS. Ptk7 and Mcc, unfancied components in non-canonical Wnt signaling and cancer. Cancers. 2016;8:68.
Tian X, Yan L, Zhang D, Guan X, Dong B, Zhao M, et al. PTK7 overexpression in colorectal tumors: clinicopathological correlation and prognosis relevance. Oncol Rep. 2016;36:1829–36.
Lhoumeau AC, Martinez S, Boher JM, Monges G, Castellano R, Goubard A, et al. Overexpression of the promigratory and prometastatic PTK7 receptor is associated with an adverse clinical outcome in colorectal cancer. PLoS One. 2015;10:e0123768.
Gillis WQ, Kirmizitas A, Iwasaki Y, Ki DH, Wyrick JM, Thomsen GH. Gtpbp2 is a positive regulator of Wnt signaling and maintains low levels of the Wnt negative regulator Axin. Cell Commun Signal. 2016;14:15.
Takahashi M, Fujita M, Furukawa Y, Hamamoto R, Shimokawa T, Miwa N, et al. Isolation of a novel human gene, APCDD1, as a direct target of the beta-Catenin/T-cell factor 4 complex with probable involvement in colorectal carcinogenesis. Cancer Res. 2002;62:5651–6.
Mori Y, Olaru AV, Cheng Y, Agarwal R, Yang J, Luvsanjav D, et al. Novel candidate colorectal cancer biomarkers identified by methylation microarray-based scanning. Endocr Relat Cancer. 2011;18:465–78.
Kim SS, Shago M, Kaustov L, Boutros PC, Clendening JW, Sheng Y, et al. CUL7 is a novel antiapoptotic oncogene. Cancer Res. 2007;67:9616–22.
Zhi J, Sun J, Wang Z, Ding W. Support vector machine classifier for prediction of the metastasis of colorectal cancer. Int J Mol Med. 2018;41:1419–26.
Shigeyasu K, Okugawa Y, Toden S, Boland CR, Goel A. Exportin-5 functions as an oncogene and a potential therapeutic target in colorectal cancer. Clin Cancer Res. 2017;23:1312–22.
Bera TK, Zimonjic DB, Popescu NC, Sathyanarayana BK, Kumar V, Lee B, et al. POTE, a highly homologous gene family located on numerous chromosomes and expressed in prostate, ovary, testis, placenta, and prostate cancer. Proc Natl Acad Sci USA. 2002;99:16975–80.
Bera TK, Saint Fleur A, Lee Y, Kydd A, Hahn Y, Popescu NC, et al. POTE paralogs are induced and differentially expressed in many cancers. Cancer Res. 2006;66:52–6.
Zhou L, Xu Z, Ren X, Chen K, Xin S. MicroRNA-124 (MiR-124) inhibits cell proliferation, metastasis and invasion in colorectal cancer by downregulating Rho-associated protein kinase 1(ROCK1). Cell Physiol Biochem. 2016;38:1785–95.
Sari I, Berberoglu B, Ozkara E, Oztuzcu S, Camci C, Demiryurek AT. Role of rho-kinase gene polymorphisms and protein expressions in colorectal cancer development. Pathobiology. 2013;80:138–45.
Usener D, Schadendorf D, Koch J, Dubel S, Eichmuller S. cTAGE: a cutaneous T cell lymphoma associated antigen family with tumor-specific splicing. J Invest Dermatol. 2003;121:198–206.
Gerhardt A, Usener D, Keese M, Sturm J, Schadendorf D, Eichmuller S. Tissue expression and sero-reactivity of tumor-specific antigens in colorectal cancer. Cancer Lett. 2004;208:197–206.
Choi MR, An CH, Yoo NJ, Lee SH. Laminin gene LAMB4 is somatically mutated and expressionally altered in gastric and colorectal cancers. APMIS. 2015;123:65–71.
Lassmann S, Weis R, Makowiec F, Roth J, Danciu M, Hopt U, et al. Array CGH identifies distinct DNA copy number profiles of oncogenes and tumor suppressor genes in chromosomal- and microsatellite-unstable sporadic colorectal carcinomas. J Mol Med. 2007;85:293–304.
Jiao X, Aravidis C, Marikkannu R, Rantala J, Picelli S, Adamovic T, et al. PHIP—a novel candidate breast cancer susceptibility locus on 6q14.1. Oncotarget. 2017;8:102769–82.
Liu W, Jiao X, Thutkawkorapin J, Mahdessian H, Lindblom A. Cancer risk susceptibility loci in a Swedish population. Oncotarget. 2017;8:110300–10.
Thutkawkorapin J, Mahdessian H, Barber T, Picelli S, von Holst S, Lundin J, et al. Two novel colorectal cancer risk loci in the region on chromosome 9q22.32. Oncotarget. 2018;9:11170–9.
Li J, Zhang N, Zhang R, Sun L, Yu W, Guo W, et al. CDC5L promotes hTERT expression and colorectal tumor growth. Cell Physiol Biochem. 2017;41:2475–88.
Acknowledgements
We thank all patients for their contribution. Genotyping and sequencing were performed by the SNP&SEQ Technology Platform and the Genomics Production Facility of National Genomics Infrastructure (NGI), hosted by Science for Life Laboratory in Uppsala and Stockholm, Sweden. We acknowledge The Swedish Twin Registry for access to data. The Swedish Twin Registry is managed by Karolinska Institutet and receives funding through the Swedish Research Council under the grant no. 2017-00641. Colorectal Transdisciplinary Study (CORECT): The content of this manuscript does not necessarily reflect the views or policies of the National Cancer Institute or any of the collaborating centers in the CORECT Consortium, nor does mention of trade names, commercial products or organizations imply endorsement by the US Government or the CORECT Consortium.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
von Holst, S., Jiao, X., Liu, W. et al. Linkage analysis revealed risk loci on 6p21 and 18p11.2-q11.2 in familial colon and rectal cancer, respectively. Eur J Hum Genet 27, 1286–1295 (2019). https://doi.org/10.1038/s41431-019-0388-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-019-0388-3
