
Abstract
Predicting metabolizer phenotype (MP) is typically performed using data from a single gene. Cytochrome p450 family 2 subfamily D polypeptide 6 (CYP2D6) is considered the primary gene for predicting MP in reference to approximately 30% of marketed drugs and endogenous toxins. CYP2D6 predictions have proven clinically effective but also have well-documented inaccuracies due to relatively high genotype–phenotype discordance in certain populations. Herein, a pathway-driven predictive model employs genetic data from uridine diphosphate glucuronosyltransferase, family 1, polypeptide B7 (UGT2B7), adenosine triphosphate (ATP)-binding cassette, subfamily B, number 1 (ABCB1), opioid receptor mu 1 (OPRM1), and catechol-O-methyltransferase (COMT) to predict the tramadol to primary metabolite ratio (T:M1) and the resulting toxicologically inferred MP (t-MP). These data were then combined with CYP2D6 data to evaluate performance of a fully combinatorial model relative to CYP2D6 alone. These data identify UGT2B7 as a potentially significant explanatory marker for T:M1 variability in a population of tramadol-exposed individuals of Finnish ancestry. Supervised machine learning and feature selection were used to demonstrate that a set of 16 loci from 5 genes can predict t-MP with over 90% accuracy, depending on t-MP category and algorithm, which was significantly greater than predictions made by CYP2D6 alone.
Similar content being viewed by others


Introduction
Pharmacogenetic studies typically rely on targeted monogenic genotyping approaches (i.e., detection of targeted single-nucleotide variants (SNVs) from one gene) to characterize the way populations or individuals respond to drugs [1,2,3,4]. The cytochrome p450 family 2, subfamily D, polypeptide 6 (CYP2D6) locus is a gene routinely used to predict metabolism of various compounds due to its involvement in phase I metabolism of approximately 30% of marketed drugs and endogenous toxins [3, 5,6,7]. CYP2D6 genotype–phenotype correlations have demonstrated relatively high efficacy in various clinical applications, however, notable genotype–phenotype discordance is documented [8,9,10]. It is understood that drug ADME-R (absorption, distribution, metabolism, excretion, and response) are dependent upon protein pathways, not the activity of a single protein. Consequently, one-gene one-phenotype predictive models do not utilize extended ADME-R information. Altar et al. demonstrated that combinatorial approaches (i.e., genetic data from multiple proteins) to predicting metabolizer phenotype (MP) have significantly more efficacious patient outcomes when compared to a single-gene single-phenotype model for psychiatric compounds [11, 12]. Pathway-driven pharmacogenetic studies have been performed in relatively few drug classes and typically utilize relatively few loci (i.e., genes or SNVs) [13,14,15], but the success of this type of model has not been evaluated for extended metabolic pathways (e.g., one or two genes versus multiple genes representing various stages of ADME-R).
CYP2D6 is readily implicated in O-demethylation of tramadol to form O-desmethyltramadol (M1). Tramadol is among the most widely prescribed opioid analgesics in the United States and as such contributes to the critical public health opioid usage and distribution crisis [16, 17]. Given the overwhelmingly high tramadol-related deaths in the United States (over 19,000; see https://www.kff.org/other/state-indicator/opioid-overdose-deaths-by-type-of-opioid), it is essential that predictive models include as much data as possible to address the degree of CYP2D6 genotype–phenotype discordance observed in individuals and in some populations. It is reasonable to hypothesize that a combinatorial predictive model of tramadol metabolism using genetic information from proteins representative of phase II metabolism, active metabolite distribution, and neurotransmitter and analgesia propagation would provide a more complete picture of how an individual responds to tramadol.
Supervised machine learning identifies underlying relationships describing the interaction between a known outcome variable (i.e., MP) and highly dimensional explanatory variables (i.e., genotypes). To our knowledge, machine learning is not readily used to identify loci for predicting patient MP but may offer considerable advantages for pathway-driven pharmacogenetic analyses via feature selection. Herein, a pathway-driven predictive model of tramadol ADME-R was evaluated to identify features (i.e., single nucleotide [SNPs] and/or insertion/deletion polymorphisms) capable of classifying members of a deceased, tramadol-exposed Finnish population into toxicologically inferred MP (t-MP) categories. The genetic data from uridine diphosphate glucuronosyltransferase, family 1, polypeptide B7 (UGT2B7), adenosine triphosphate (ATP)-binding cassette, subfamily B, number 1 (ABCB1), opioid receptor mu 1 (OPRM1), and catechol-O-methyltransferase (COMT) were used in combination with CYP2D6 data [18] and demonstrate increased prediction accuracy and correlation coefficients for the t-MP and T:M1 outcome variables, respectively. These predictions were made using a substantially reduced number of loci (16 and 33 for t-MP and T:M1, respectively) offering promise for design and clinical implementation of accurate and reproducible tramadol response models.
Subjects and methods
Subjects
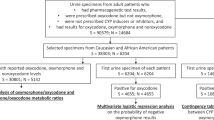
A total of 208 DNA samples from deceased, tramadol-exposed individuals of Finnish ancestry were used in this study. Samples were collected in Finland between 2001 and 2012 according to the ethical handling of human subjects policies at the University of Helsinki and transferred to the University of North Texas Health Science Center (Institutional Review Board protocol 2016-051). Samples were stored on Whatman® FTA® cards (GE Healthcare Life Science, Marlborough, MA, USA) [19]. Each sample was accompanied by comprehensive toxicology results including measurements for over 100 routinely tested compounds, primary and secondary cause of death (CoD), manner of death (MoD), age, and sex. Sample inclusion was determined based on toxicological detection of tramadol and its primary metabolite O-desmethyltramadol. Using analysis of variance, there were no detectable relationships between the presence/absence of these compounds or the concentration of each compound and T:M1 ratio used to classify each subject. Model-based clustering of each sample was performed in R using the mclust package (version 5.2.3) [20] and the T:M1 ratio for each sample. Additional information on sample collection, toxicological analyses, and DNA extraction and quantitation were described by Wendt et al. [18].
Marker selection, library preparation, and massively parallel sequencing
A TruSeq® Custom Amplicon (TSCA) Low Input library preparation panel (Illumina®, Inc.) was designed using the Illumina DesignStudio™ (see https://www.illumina.com/informatics/sample-experiment-management/custom-assay-design.html; accessed June 2017). The exons of four pharmacogenes (UGT2B7, ABCB1, OPRM1, and COMT) were targeted for kit design (Table 1). These genes were selected based on previous work highlighting their potential to predict opioid response outcomes [13, 14, 21].
Library preparation was performed using 10 ng of genomic DNA and followed the manufacturer’s recommended protocol. Two modifications were made to the TSCA Low Input protocol: (1) during the Remove Unbound Oligos step, sample purification beads were allowed to dry for 1 min instead of the indicated 5 min; and (2) prior to library cleanup, the hybridization plate was placed on a magnetic stand for 2 min before 45 μL of supernatant were transferred to the cleanup plate. Cleaned-up library traces were spot-checked using the Agilent 2200 TapeStation (Agilent Technologies, Waldbronn, Germany) using the Agilent 2200 High Sensitivity D1000 ScreenTape System according to the manufacturer’s recommended protocol (see https://support.illumina.com/content/dam/illumina-support/documents/documentation/chemistry_documentation/samplepreps_truseq/truseqcustomamplicon/truseq-custom-amplicon-low-input-reference-guide-1000000002191–04.pdf; accessed July 2017). Sample libraries were normalized and pooled in batches of 32 and sequenced on the MiSeq (Illumina) using the MiSeq Reagent kit v2 (500 cycles) with 2 × 250 bp read length.
Alignment, variant analysis, and machine learning
Resulting .fastq files were locally aligned to the hg19/GRCh37 reference genome using the Burrows-Wheeler Aligner and SamTools [22,23,24]. Variant calling was performed in Genome Analysis ToolKit (GATK) [25]. Resulting .vcf files were, or were converted to, standard input for VCFtools [26], Genome-wide Complex Trait Analysis [27], PLINK [28], IMPUTE2 v2.3.2 [29], and various in-house Excel-based workbooks. Variants may be accessed from ClinVar under submission number SUB4785168.
Supervised machine learning was performed in the Waikato Environment for Knowledge Analysis as described previously [18, 30, 31] using four classifiers: regularized multinomial logistic regression (RMLR; t-MP only), 1-nearest neighbor (1NN; t-MP and T:M1), random forest (RF; t-MP and T:M1), and linear regression (LR; T:M1 only). Feature selection and leave-one-out cross-validation were used to reduce the size of the model and assess model accuracy, respectively. Note that leave-one-out cross-validation also minimizes the effects of overfitting the model by training a given model in the absence of one sample and evaluating model accuracy on the left-out sample. Unless otherwise stated, sample n − 1 (i.e., 207-fold) cross-validation was performed.
Results
Samples
The cohort in this study represents a larger sampling of deceased tramadol-exposed individuals of Finnish ancestry than reported previously [18]. International Classification of Diseases, Tenth Revision (ICD-10) CoD codes (see International Statistical Classification of Diseases and Related Health Problems Tenth Revision [ICD-10]-WHO Version for 2016. http://apps.who.int/classifications/icd10/browse/2016/en#/I; accessed 21 Apr 2017) and a more generalized manner of death (MoD) (e.g., disease, trauma, suicide, medical treatment, or undetermined) were assigned to each individual at the time of medico-legal autopsy (Table 2) The average measured concentrations of tramadol and M1 and T:M1 ratio were 4.04 mg/l ± 5.94, 0.447 ± 0.769 mg/l, and 11.6 ± 18.3, respectively. There was no significant difference in mean T:M1 between males (12.8 ± 25.0; N = 127; mean age 52.2 years ± 17.9) and females (10.9 ± 12.3; N = 81; mean age 60.0 years ± 18.3).
Assuming unequal variance, five clusters were identified, using the R package mclust [20], and used to sort each sample into a t-MP category based on the following thresholds: poor metabolizers (PM; T:M1 ≥ 50; N = 5), intermediate metabolizers (IM; 50 > T:M1 ≥ 20; N = 20), slow normal metabolizers (NM-S; 20 > T:M1 ≥ 8; N = 67), fast normal metabolizers (NM-F; 8 > T:M1 ≥ 3; N = 91), and ultra-rapid metabolizers (UM; 3 > T:M1 ≥ 1; N = 25).
Library preparation panel and sequencing performance
The massively parallel sequencing (MPS) panel targeted 216 exonic amplicons with a probe-based chemistry (Tables S1 and S2). Based on requirements for probe placement, some intronic regions also were obtained. The average amplicon length was 177 bases ± 6.84. Note that ABCB1 and COMT had small gaps after panel design (Table 1) resulting in lack of genotype data for two exonic SNPs observed once in the Exome Aggregation Consortium and 1000 Genomes Project databases (1kGP) [32]. Variant effect prediction indicated possibly damaging consequences of ABCB1 gap variant NC_000007.13:g.87145971C>G (SIFT: 0; PolyPhen-v2: 0.995) [33, 34].
Single-nucleotide variants
After application of a 10× locus read-depth threshold, the average alternate allele frequency of 8546 SNVs was 0.0300 ± 0.0876. After Bonferroni correction (padj_heterozygous_loci = 3.59 × 10–5; ~70 deviations expected due to chance alone), 8 loci significantly deviated from expectations of Hardy-Weinberg equilibrium, all of which exhibited significant excess heterozygosity in this Finnish population, suggesting relatively little population substructure in this cohort.
After correction for multiple testing (padj = 2.42 × 10−6), 14 SNVs were significantly associated with the rate of tramadol O-demethylation (Fig. 1a and Table 3). Two pairs of loci exhibited significant linkage disequillibria (NC_000007.13:g.87229006T > G/NC_000022.10:g.19938432G > A [N = 164 and 147 genotypes, respectively] and NC_000004.11:g.69972849T > C/NC_000022.10:g.19938432G>A [N = 150 and 147 genotypes, respectively]).

a Association between the tramadol to O-desmethyltramadol ratio (T:M1) for individual genotypes at UGT2B7, ABCB1, OPRM1, and COMT. A dashed horizontal line indicates the threshold for significance after correction for multiple testing (−log10(padj) = 5.62); loci are labeled if they exceed the significance threshold and have regression coefficients ≥ 0.45 (arbitrarily selected to avoid locus label overlap). b Heritability summary of restricted maximum likelihood (--reml in Genome-wide Complex Trait Analysis) analyses, with the first 20 eigenvectors as covariates, of the toxicologically inferred metabolizer phenotype (t-MP; solid gray triangles) and the associated t-MP (T:M1; solid black circles) phenotypes. A dashed horizontal line indicates the threshold for significance after correction for multiple testing (−log10(padj) = 2.79); two data points are labeled, indicating that the individual (UGT2B7) and combined (CYP2D6/UGT2B7) genotype information significantly explained a relatively high proportion of phenotypic variance
The SNP heritability (h2) of t-MP and T:M1 was inferred using the --reml command in GATK with individual and pairwise combinations of two, three, four, and all five genes (Fig. 1b). In general, the variability of t-MP was poorly explained regardless of gene or gene combination. However, after correction for multiple testing (padj = 0.00161), the SNVs from UGT2B7 (h2T:M1 = 0.821; p = 1.22 × 10−6) and the combination of SNVs from CYP2D6/UGT2B7 (h2T:M1 = 0.786; p = 4.04 × 10−4) significantly explained relatively large proportions of the variation in T:M1 with relatively little error (0.0594 and 0.0758 for UGT2B7 and CYP2D6/UGT2B7, respectively).
Predicting t-MP
Predictions of t-MP and T:M1 were performed in two phases: (1) classification of MP using combined unphased genotype data from UGT2B7, ABCB1, OPRM1, and COMT; and (2) classification with computationally phased genotypes from the same four genes. In general, unphased genotypes predicted t-MP and T:M1 variables with less accuracy and lower correlation coefficients than the phased genotype models. The results presented herein focus on predictions of t-MP and T:M1 using phased genotype data.
The 208 tramadol-exposed individuals of Finnish ancestry used for classification represent five classes of t-MP. Using three supervised machine learning models, t-MP prediction accuracies were relatively low with mean accuracies of 19.2% ± 39.7 (RF), 20.4% ± 23.0 (1NN), and 25.2% ± 15.6 (RMLR), which are not better than random chance (20.9%, 10%-trimmed mean). These accuracies represent poor prediction of all five t-MPs, with lack of a true positive prediction for the t-IM and t-PM categories in all three models. Overall, the RMLR classifier predicted t-MP with significantly higher accuracies than the 1NN or RF classifiers (p < 0.001).
Feature selection was used to evaluate classification accuracies as a reduced number of SNVs are provided for each model. The models were evaluated with features used in >0, >12, >25, >50, and >75% of cross-validation folds (Fig. 2). Classification accuracies generally increased for all five t-MP categories with the RMLR classifier outperforming the LR and 1NN models. RMLR predicted the t-MP variable with mean accuracies for t-UM, t-NM-F, t-NM-S, and t-IM that were 1.22- (25.6% ± 2.19), 3.35- (70.1% ± 2.38), 2.64- (55.2% ± 4.47) and 2.01-fold (42.0% ± 2.74) greater than random chance (20.9%; 10%-trimmed mean), respectively. Note that t-PMs were not reliably predicted with any algorithm.

Summary of machine learning classification accuracies for four metabolizer phenotype clusters (t-UM ultra-rapid, t-NM-F normal/extensive [fast], t-NM-S normal/extensive [slow], t-IM intermediate) using phased UGT2B7, ABCB1, OPRM1, and COMT data aligned to the hg19/GRCh37 reference genome for varying feature selection stringencies (features used in greater than 0, 12, 25, 50, and 75% of cross-validation folds) compared to the accuracy of the model using all genotype data from UGT2B7, ABCB1, OPRM1, and COMT. Three machine learning algorithms are depicted: 1-nearest neighbor (1NN), random forest (RF), and regularized multinomial logistic regression (RMLR); dashed lines represent the average predictive accuracy due to random chance (20.9%; 10%-trimmed mean)
The RMLR classifier relies on regularization to reduce model complexity after proportionally penalizing the squared effect of each feature on the outcome. For this classifier, ridge regularization parameters ranging from 1 × 10−10 to 0.01 were evaluated. The mean root mean squared error (RMSE) of these assessments changed negligibly with modified ridge parameter (mean RMSE 0.408 ± 0.007) while model accuracy increased (maximum number of correctly classified individuals was 65.4%). Note that while feature selection provided no notable increase in t-PM classification accuracy, reducing RMLR model complexity (ridge parameter ≤1 × 10−4) increased t-PM classification accuracy to 20%.
Wendt et al. [18] previously described t-MP classification using CYP2D6 alone. That study analyzed a subset of the current sample set (N = 44/208 individuals), which had genetic data for a fully comprehensive t-MP prediction. Note that because of limited sampling, this cohort represented only four MP categories (i.e., t-PMs were not observed) and used 43-fold cross-validation. Using a comprehensive, pathway-driven model with all 10,421 SNVs from CYP2D6 [18], UGT2B7, ABCB1, OPRM1, and COMT, accuracy of the RF, 1NN, and RMLR classifiers modestly increased to 27.3%, 27.3%, and 20.5%, respectively. Feature selection increased these classification accuracies with the RMLR classifier again outperforming the 1NN and RF models in overall accuracy (mean of 60.6% ± 19.1 overall; 61.5% for t-UM, 75.0% for t-NM-F, 72.7% for t-NM-S, and 33.3% for t-IM; Fig. 3a). Relative to the CYP2D6 predictions, the pathway-driven model using CYP2D6, UGT2B7, ABCB1, OPRM1, and COMT together provided significantly higher classification accuracies for t-MP (p = 0.0190; paired t-test). The maximum classification accuracy reached 93.8% using only 16 SNVs.

Summary of machine learning classification accuracies for metabolizer phenotype (t-UM ultra-rapid; t-NM normal/extensive, t-IM intermediate; a) and the tramadol to O-desmethyltramadol ratio (T:M1; b) using phased CYP2D6 [18], UGT2B7, ABCB1, OPRM1, and COMT data aligned to the hg19/GRCh37 reference relative to using CYP2D6 alone. In a, varying feature selection stringencies (i.e., features used in greater than 0, 12, 25, 50, and 75% of cross-validation folds) and supersized machine learning algorithms (i.e., 1-nearest neighbor (1NN), random forest (RF), and regularized multinomial logistic regression (RMLR)) were used; dashed lines represent the average predictive accuracy due to random chance (39.9% for pathway model and 23.8% for CYP2D6 model; 10%-trimmed mean). Note the cube root scale in b with standard error shown in gray shading along the length of each robust linear regression
Predicting T:M1
Supervised machine learning was performed on the T:M1 outcome variable in the same manner as t-MP except that the LR classifier was used instead of RMLR. The average difference between actual and predicted T:M1 (ΔT:M1) using 8546 phased SNVs from UGT2B7, ABCB1, OPRM1, and COMT was −1.54 ± 17.6, indicating overall underestimation of T:M1. The 1NN classifier underestimated T:M1 with significantly greater magnitude than the RF and LR classifiers (p < 1 × 10−20) with mean ΔT:M1 of −5.89 ± 18.9 (1NN), 0.191 ± 19.0 (LR), and −0.581 ± 18.8 (RF).
The same five feature inclusion thresholds were evaluated for the T:M1 variable (Fig. 4). Overall, T:M1 was modestly predicted regardless of feature inclusion stringency or supervised machine learning algorithm used. The average correlation coefficients between actual and predicted T:M1 were not significantly different regardless of feature inclusion stringency; however, the correlation coefficients from the LR classifier (r2 = 0.113 ± 0.0212) were significantly lower than those of the 1NN (r2 = 0.277 ± 0.0520) and RF (r2 = 0.284 ± 0.0307) classifiers. The maximum observed correlation coefficient was 0.383 with the 1NN classifier.

Summary of tramadol O-demethylation (T:M1 ratio) predictions using three supervised machine learning algorithms (1-nearest neighbor [1NN], linear regression [LR], and random forest [RF]) and five feature inclusion criteria (i.e., features included in >0, >12, >25, >50, and >75% of cross-validation folds) relative to the prediction using genotype data from 8546 loci in UGT2B7, OPRM1, ABCB1, and COMT for phased and unphased hg19/GRCh37 data. The dashed lines represent the average predictive accuracy due to random chance (0.112; 10%-trimmed mean). The individual data points contributing to these correlation coefficients are shown in Figure S1
T:M1 predictions with CYP2D6 alone and the pathway-driven model were evaluated. For 44 samples and the full set of genotype data without feature selection (NSNVs = 10,421), the pathway-driven model (mean ΔT:M1 = –1.35 ± 12.8) yielded a significantly lower ΔT:M1 than the CYP2D6 model (mean ΔT:M1 = −6.79 ± 34.9; p = 0.0293). This observation was especially true for the LR classifier (ΔT:M1pathway = −0.101 ± 12.3 and ΔT:M1CYP2D6 = −20.1 ± 56.2), which had a significantly decreased ΔT:M1 with the pathway-driven model (p < 0.01). Note that the 1NN classifier performed less well with pathway-driven data (ΔT:M1 = −4.40 ± 13.6) relative to the CYP2D6 data (ΔT:M1 = −1.10 ± 14.3). This observation holds true for the LR classifier using the >12, >25, >50, and >75% feature inclusion thresholds with significant improvements of the pathway-driven models relative to the equivalent feature inclusion model using CYP2D6 alone (p < 0.00330).
Discussion
This study evaluated a combinatorial, pathway-driven pharmacogenetic model of tramadol O-demethylation using a custom TSCA Low Input library preparation panel targeting the exons of UGT2B7, ABCB1, OPRM1, and COMT, which are responsible for various stages of tramadol ADME-R. Note that while exons were targeted for panel design, optimization of amplicon size involved capturing some intronic loci. While not specifically interrogated here, there are common and intronic, promoter, enhancer, and/or silencer SNVs that have been implicated in variable gene expression, splicing, and/or post-translational modifications that need to be explored further to develop fully pathway-driven models of drug-specific and drug-class ADME-R [35]. Utilizing comprehensive MPS (referred to as next-generation sequencing by Ingelman-Sundberg et al. [35]) to detect and incorporate rare variants could be valuable for increasing ADME-R prediction accuracies, which is supported by the work presented here.
Fourteen SNVs were significantly associated with T:M1; however, only one of these (NC_000007.13:g.87178819C>T) was predicted to alter protein function by causing a valine to isoleucine amino-acid change adjacent to the ATP-binding cassette signature sequence, between the signature and Q-loop. This locus was observed with an allele frequency of 0.000200 in the 1kGP (South Asian super-population only) but was not present in the Sequencing Initiative Suomi (SISu; Finnish population only; see Sequencing Initiative Suomi project (SISu), Institute for Molecular Medicine Finland, University of Helsinki, Finland. URL: http://sisuproject.fi [SISu v4.1, May, 2018]). Combined Annotation Dependent Depletion indicates this locus as one of the top 0.1–1% most deleterious substitutions in the human genome with SIFT and PolyPhen-v2 providing agreeable predictions [33, 34, 36]. Its position within the binding pocket of ABCB1 is quite close to crosslinked regions of the domain; though charge is not disrupted by the change of valine to isoleucine, the extra methyl group of isoleucine may sterically hinder appropriate crosslinking between nucleotide binding domains 1 and 2 [37,38,39].
The t-MP variable was relatively poorly explained by the genetic data from CYP2D6, UGT2B7, ABCB1, OPRM1, and COMT (10,421 SNVs) with restricted maximum likelihood analysis for all combinations of genes (i.e., singlets, pairs, trios, etc.). Interestingly, T:M1 was highly explained by genetic variation at UGT2B7 and the combination of UGT2B7-CYP2D6, which was likely an artifactual inflation of the poor explanation of T:M1 by CYP2D6 relative to UGT2B7. This is an interesting observation since CYP2D6 data alone are routinely relied upon for predicting drug response. CYP2D6 information is typically used as a predictor for the ratio of drug concentration to active metabolite concentration [1, 4, 5, 40, 41], thereby guiding safer first-pass drug doses in place of solely trial-and-error. While CYP2D6 is considered a front-end indicator of drug to metabolite ratio, UGT2B7 is responsible for efficient glucuronidation of the metabolite, facilitating its biliary excretion. Increased or decreased activity of UGT2B7 may be associated with fast or slow excretion of a metabolite, respectively, influencing patient outcomes (i.e., slow glucuronidation of morphine via UGT2B7 may result in accumulation of morphine and its associated toxicity). The data herein suggest that UGT2B7 may serve as an equally, or more, informative back-end indicator of the same phenotype predicted frequently by CYP2D6.
Three supervised machine learning algorithms were used to predict t-MP and T:M1. In general, t-MP was predicted well using the combination of UGT2B7, ABCB1, OPRM1, and COMT genotype information. Classification accuracies were especially high for the t-NM-F and t-NM-S categories, demonstrating reliable detection of normal versus non-normal metabolizers. Predicting the direction of non-normality was less successful; however, the highest feature inclusion stringency increased classification accuracies of the t-UM, t-IM, and t-PM categories. Overall, t-PM was poorly predicted with these SNVs using all three models. It is important to note that this MP is regularly characterized by a variety of structural aberrations and while SNV predictions may have been poor, large cohorts of known PMs may enable SNV models in the absence of structural information. T:M1 was poorly predicted using the three selected algorithms; however, predicting this outcome variable was noticeably, and significantly, algorithm-dependent. While using multiple machine learning algorithms and feature inclusion criteria may be seen as biased, this approach may be beneficial for future application and possible best practices for clinical implementation of predictive models based on genotyping approaches. The data presented using a pathway-driven predictive model of tramadol ADME-R demonstrated clear algorithm differences with the RF and 1NN classifiers exhibiting the highest correlation coefficients between predicted and actual T:M1. While the RF classifier produced some of the highest t-MP prediction accuracies, it failed to reliably predict the extreme categories (i.e., t-PM, t-IM, and t-UM); on the other hand, the RMLR classifier predicted t-MP quite well across a range of categories.
A subset of samples were assayed in a truly combinatorial fashion, with full genotype data for CYP2D6, UGT2B7, ABCB1, OPRM1, and COMT. By applying the RMLR, 1NN, RF, and LR classifiers to this subset of samples, it was demonstrated that a pathway-driven model of tramadol ADME-R more accurately predicts outcome than a CYP2D6-driven model. The predictive models presented here for T:M1 and t-MP achieved high accuracies depending on category and algorithm used; however, the power of this approach is represented by the number of loci used to predict either outcome variable. Relative to a model using 10,421 SNVs (maximum accuracy of 68.8%), t-MP can be predicted with up to 93.8% accuracy using 16 SNVs (651-fold reduction in SNVs) and T:M1 can be predicted with a correlation coefficient up to 0.329 with 33 SNVs (316-fold reduction in SNVs; Table 4). While it is possible that large feature reduction facilitated overfitting, leave-one-out cross-validation and model-dependent regularization were used to reduce these effects.
The current clinical standard of CYP2D6 genotyping has inaccuracies in multiple ancestral populations and/or diseased populations, which in itself warrants exploration of alternative or additional predictions of phenotype. We have demonstrated that the performance of a pathway-driven predictive model of tramadol ADME-R, using rare and common genetic variants, was relatively low; however, utilizing pathway information may outperform the current clinical standard. It is likely that a more broadly applicable predictive model will employ additional DNA elements (i.e., introns, promoters, enhancers, silencers, distant regulatory elements, etc.), including common and rare variants. The data described here demonstrate the feasibility of pathway-driven models and serve as a foundation to build clinically applicable assays relying on protein pathway information rather than single-gene predictions of complex drug metabolism phenotypes. Additional DNA elements could be explored for development of relatively small, accurate, and easily implemented MPS library preparation panels for clinical application of drug response models.
References
Gaedigk A, Sangkuhl K, Whirl-Carrillo M, Klein T, Leeder JS. Prediction of CYP2D6 phenotype from genotype across world populations. Genet Med. 2017;19:69–76.
Ingelman-Sundberg M. Genetic polymorphisms of cytochrome P450 2D6 (CYP2D6): clinical consequences, evolutionary aspects and functional diversity. Pharm J. 2005;5:6–13.
Leppert W. CYP2D6 in the metabolism of opioids for mild to moderate pain. Pharmacology. 2011;87:274–85.
Sistonen J, Fuselli S, Palo JU, Chauhan N, Padh H, Sajantila A. Pharmacogenetic variation at CYP2C9, CYP2C19, and CYP2D6 at global and microgeographic scales. Pharmacogenet Genomics. 2009;19:170–9.
Koski A, Sistonen J, Ojanpera I, Gergov M, Vuori E, Sajantila A. CYP2D6 and CYP2C19 genotypes and amitriptyline metabolite ratios in a series of medicolegal autopsies. Forensic Sci Int. 2006;158:177–83.
Levo A, Koski A, Ojanpera I, Vuori E, Sajantila A. Post-mortem SNP analysis of CYP2D6 gene reveals correlation between genotype and opioid drug (tramadol) metabolite ratios in blood. Forensic Sci Int. 2003;135:9–15.
Mas S, Gasso P, Torra M, Bioque M, Lobo A, Gonzalez-Pinto A, et al. Intuitive pharmacogenetic dosing of risperidone according to CYP2D6 phenotype extrapolated from genotype in a cohort of first episode psychosis patients. Eur Neuropsychopharmacol. 2017;27:647–56.
De Andres F, Teran S, Hernandez F, Teran E, LL A. To genotype or phenotype for personalized medicine? CYP450 drug metabolizing enzyme genotype-phenotype concordance and discordance in the ecuadorian population. OMICS. 2016;20:699–710.
Gaedigk A, Bradford LD, Marcucci KA, Leeder JS. Unique CYP2D6 activity distribution and genotype-phenotype discordance in black Americans. Clin Pharmacol Ther. 2002;72:76–89.
Shiran MR, Chowdry J, Rostami-Hodjegan A, Ellis SW, Lennard MS, Iqbal MZ, et al. A discordance between cytochrome P450 2D6 genotype and phenotype in patients undergoing methadone maintenance treatment. Br J Clin Pharmacol. 2003;56:220–4.
Altar CA, Carhart J, Allen JD, Hall-Flavin D, Winner J, Dechairo B. Clinical utility of combinatorial pharmacogenomics-guided antidepressant therapy: evidence from three clinical studies. Mol Neuropsychiatry. 2015;1:145–55.
Altar CA, Carhart JM, Allen JD, Hall-Flavin DK, Dechairo BM, Winner JG. Clinical validity: combinatorial pharmacogenomics predicts antidepressant responses and healthcare utilizations better than single gene phenotypes. Pharm J. 2015;15:443–51.
Baber M, Chaudhry S, Kelly L, Ross C, Carleton B, Berger H, et al. The pharmacogenetics of codeine pain relief in the postpartum period. Pharm J. 2015;15:430–5.
Bastami S, Gupta A, Zackrisson AL, Ahlner J, Osman A, Uppugunduri S. Influence of UGT2B7, OPRM1 and ABCB1 gene polymorphisms on postoperative morphine consumption. Basic Clin Pharmacol Toxicol. 2014;115:423–31.
Sistonen J, Madadi P, Ross CJ, Yazdanpanah M, Lee JW, Landsmeer ML, et al. Prediction of codeine toxicity in infants and their mothers using a novel combination of maternal genetic markers. Clin Pharmacol Ther. 2012;91:692–9.
Seya MJ, Gelders SF, Achara OU, Milani B, Scholten WK. A first comparison between the consumption of and the need for opioid analgesics at country, regional, and global levels. J Pain Palliat Care Pharmacother. 2011;25:6–18.
Solanki DR, Koyyalagunta D, Shah RV, Silverman SM, Manchikanti L. Monitoring opioid adherence in chronic pain patients: assessment of risk of substance misuse. Pain Physician. 2011;14:E119–31.
Wendt FR, Novroski NMM, Rahikainen AL, Sajantila A, Budowle B. Supervised classification of CYP2D6 genotype and metabolizer phenotype with postmortem tramadol-exposed Finns. Am J Forensic Med Pathol. 2019;40:8–18.
Rahikainen AL, Palo JU, de Leeuw W, Budowle B, Sajantila A. DNA quality and quantity from up to 16 years old post-mortem blood stored on FTA cards. Forensic Sci Int. 2016;261:148–53.
Scrucca L, Fop M, Murphy TB, Raftery AE. mclust 5: clustering, classification and density estimation using gaussian finite mixture models. R J. 2016;8:289–317.
Owusu Obeng A, Hamadeh I, Smith M. Review of opioid pharmacogenetics and considerations for pain management. Pharmacotherapy. 2017;37:1105–21.
Li H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics. 2011;27:2987–93.
Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–9.
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–303.
Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, et al. The variant call format and VCFtools. Bioinformatics. 2011;27:2156–8.
Yang J, Lee SH, Goddard ME, Visscher PM. GCTA: a tool for genome-wide complex trait analysis. Am J Hum Genet. 2011;88:76–82.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–75.
Howie BN, Donnelly P, Marchini J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet. 2009;5:e1000529.
Schmedes SE, Woerner AE, Budowle B. Forensic human identification using skin microbiomes. Appl Environ Microbiol. 2017.
Schmedes SE, Woerner AE, Novroski NMM, Wendt FR, King JL, Stephens KM, et al. Targeted sequencing of clade-specific markers from skin microbiomes for forensic human identification. Forensic Sci Int Genet. 2018;32:50–61.
Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, et al. A global reference for human genetic variation. Nature. 2015;526:68–74.
McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GR, Thormann A, et al. The Ensembl Variant Effect Predictor. Genome Biol. 2016;17:122.
McLaren W, Pritchard B, Rios D, Chen Y, Flicek P, Cunningham F. Deriving the consequences of genomic variants with the Ensembl API and SNP Effect Predictor. Bioinformatics. 2010;26:2069–70.
Ingelman-Sundberg M, Mkrtchian S, Zhou Y, Lauschke VM. Integrating rare genetic variants into pharmacogenetic drug response predictions. Hum Genomics. 2018;12:26.
Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46:310–5.
Dhaliwal AK, Mohan A, Gill KS. Comparative analysis of ABCB1 reveals novel structural and functional conservation between monocots and dicots. Front Plant Sci. 2014;5:657.
Loo TW, Bartlett MC, Clarke DM. The “LSGGQ” motif in each nucleotide-binding domain of human P-glycoprotein is adjacent to the opposing walker A sequence. J Biol Chem. 2002;277:41303–6.
Sauna ZE, Ambudkar SV. About a switch: how P-glycoprotein (ABCB1) harnesses the energy of ATP binding and hydrolysis to do mechanical work. Mol Cancer Ther. 2007;6:13–23.
Gaedigk A, Simon SD, Pearce RE, Bradford LD, Kennedy MJ, Leeder JS. The CYP2D6 activity score: translating genotype information into a qualitative measure of phenotype. Clin Pharmacol Ther. 2008;83:234–42.
Sistonen J, Sajantila A, Lao O, Corander J, Barbujani G, Fuselli S. CYP2D6 worldwide genetic variation shows high frequency of altered activity variants and no continental structure. Pharmacogenet Genomics. 2007;17:93–101.
Acknowledgements
The authors thank Jenny Atanasov and Mehdi Keddache from Illumina, Inc. and Cydne Holt, John Walsh, and Kameran Wong from Verogen, Inc. for library preparation product support, and Jerry Boonyaratanakornkit and Wahaj Zuberi from Exact Diagnostics for their technical assistance generating TruSeq data. We also thank Medicinska Understödsföreningen Liv och Hälsa r.f. and Helsinki University Doctoral Programme in Population Health for support for AS and A-LR, respectively.
Funding
Support for this work was provided by the Center for Human Identification at the University of North Texas Health Science Center.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Wendt, F.R., Novroski, N.M.M., Rahikainen, AL. et al. A pathway-driven predictive model of tramadol pharmacogenetics. Eur J Hum Genet 27, 1143–1156 (2019). https://doi.org/10.1038/s41431-019-0369-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-019-0369-6
