
Abstract
Here, we describe a single patient from a consanguineous family, who suffers from developmental delay, intellectual disability, hypermetropia, moderate alternating esotropia, unsteady gait, and peripheral polyneuropathy. Brain MRI revealed basal ganglia disease. Exome analysis disclosed a homozygous variant, c.452G>C (p.(Arg151Thr)), in TID1, encoding a mitochondrial J-protein chaperone that is known for its function in assisting the Hsp70 chaperone, mortalin, in mediating the refolding of denatured protein and dissolving protein aggregates. Results from in vitro import assays showed that both wild type and c.452G>C (p.(Arg151Thr)) are efficiently imported into isolated mitochondria. However, the import rate of the c.452G>C (p.(Arg151Thr)) variant was less than that of the wild-type protein. In the second part of this study, we demonstrated, in vitro, that the disaggregation function of the mortalin/Tid1 team is compromised in the TID1 c.452G>C (p.(Arg151Thr)) variant, as its chaperone activity has a level similar to that of the non-functional H→Q HPD domain variant. The results shed light on the essential function played by Tid1 during neuronal development.
Similar content being viewed by others


Introduction
The universally conserved 70 kDa heat shock protein family (Hsp70) members orchestrate numerous cellular processes involving protein folding, protein unfolding, assembly of protein complexes, and protein degradation [1,2,3,4,5,6]. The Human Hsp70 family consists of eight members that differ in their amino acid sequence, expression level and cellular location. Six of them are found in cytosol, one is found in the lumen of the ER and one, known as mortalin (HspA9), is found in the mitochondrial matrix [7]. Mortalin is a key player in three vital proteostasis processes: protein unfolding during import, folding of newly imported or stress denatured proteins, and protein degradation. In addition, mortalin is linked to a myriad of pathological conditions [8]. Notably, in yeast, three proteins, Ssc1, Ssq1, and Ecm10, are thought to perform the proteostasis functions performed by a single protein in human mitochondria, mortalin [9, 10]. The protein folding function of Hsp70 is regulated by two co-chaperones, a J-domain-containing protein (DnaJ, Mdj1, or DjA1) and the nucleotide exchange factor GrpE (Mge1). Substrate specificity is conferred by J-domain proteins, which efficiently stimulate Hsp70’s intrinsically weak ATPase activity and thereby facilitate its interactions with client proteins [11, 12]. GrpE acts as a nucleotide exchange factor to accelerate release of the substrate protein, which has presumably been refolded to its native state [13, 14].
Mortalin shares high identity of sequence and structural similarity with other members of the Hsp70 family including E. coli DnaK (51%) and Saccharomyces cerevisiae Ssc1 (65%) [15, 16]. Similar to other Hsp70 chaperones, the proteostasis function of mortalin is modulated by a J-domain-containing co-chaperone, named Tid1 in humans (DNAJA3) and a nucleotide exchange factor, human Mge1. In humans, two splicing isoforms of Tid1 were found to be expressed Tid1L (long) and Tid1S (short) [8]. Both variants have an N-terminal mitochondrial targeting sequence as well as the signature J domain, containing a conserved HPD motif. However, they differ in their carboxy-terminal tails: Tid1L has 33 amino acids, whereas Tid1S has only six amino acids [8]. Both forms were found to mediate protein refolding in cooperation with mortalin [17].
In addition to their involvement in maintaining mitochondrial homeostasis, mortalin and Tid1 were found to be dually distributed and to have additional roles outside mitochondria. For example, an interaction of mortalin with p53 is well documented [16, 18]. This interaction leads to sequestration of p53 in the cytosol, which inhibits its ability to perform its apoptotic function [19, 20]. Consequently, mortalin has been considered a possible target for anti-cancer drugs. Similar to mortalin, Tid1 was also shown to engage in a wide variety of extra-mitochondrial interactions, most related to transcription factors, receptor tyrosine kinases, viral proteins, and tumor-suppressors [21, 22]. For both proteins, most of their interactions have been associated with pathological conditions.
Despite the involvement of Tid1 in a wide range of pathological conditions, no mitochondrial proteostasis disease has yet been linked to this protein. In the present study, we report the identification of a homozygous variant c.452G>C (p.(Arg151Thr)) in Tid1, in patients exhibiting ataxia and developmental delay. Using in vitro assays, we show that the c.452G>C (p.(Arg151Thr)) variant is less efficiently imported into mitochondria and is severely impaired in its co-chaperone function.
Materials and methods
Whole-exome analysis
Exonic sequences from the patient were enriched with the Sure Select Human All Exon 50 Mb V.4 Kit (Agilent Technologies, Santa Clara, CA). Sequences (100-bp paired-end) were generated on a HiSeq2000 (Illumina, San Diego, CA). Read alignment and variant calling were performed with DNAnexus (Palo Alto, CA) using default parameters with the human genome assembly hg19 (GRCh37) as a reference. Parental consent was given for genetic studies. The gene with dbSNP ID rs1139653 and its variant were submitted to the database at www.LOVD.nl/CAV3 with ID: 0000438528. The study was performed with the approval of the ethical committees of Hadassah Medical Center and the Israeli Ministry of Health.
Import of the Tid1 variant into mitochondria, in vitro
Tid1 wild type or c.452G>C (p.(Arg151Thr)) precursor proteins were synthesized in vitro in the presence of [35S]methionine using the TnT® Quick Coupled Transcription/Translation System (Promega). Import was carried out for 20 or 60 min at 30 °C with isolated yeast mitochondria as described previously [23]. At the indicated time points, aliquots were removed and diluted into ice-cold sucrose buffer containing 1 µM valinomycin to stop further import. The samples were then treated with proteinase K for 10 min on ice, to degrade protein that did not undergo import. An additional control sample that was not treated with proteinase K was taken at 60′ (-PK). The samples were subsequently analyzed by SDS–PAGE and autoradiography. A control import system using 1–69 of the yeast DHFR Su9 is provided for comparison. T shows the total amount of precursor that was used for each time point. Densitometry analysis was carried out using the TotalLab TL120 V2006.
Purification of recombinant Tid1L
The recombinant form of Tid1L was expressed and purified following a method described previously [24]. Briefly, E. coil BL-21 tuner strain carrying pET21d (+) plasmid, with the relevant Tid1 construct, was induced for protein over expression with 1 mM Isopropyl β-D-thiogalactopyranoside (IPTG) for 6 h at 16 °C. Tid1L was found in the pellet fraction (inclusion bodies) of the bacterial cell lysate. Previous experiments showed that 3 M urea rescues most of the Tid1L from the inclusion bodies. Therefore, we adjusted the bacterial cell lysate with 5 mM imidazole, 3 M urea and passed it through Ni-NTA agarose. Bound, his-tagged, Tid1L was eluted with 300 mM imidazole. Eluted protein was concentrated and dialyzed against 20 mM Tris, pH 7.4, 0.5 M NaCl, 10% glycerol, 0.05% Brij-35, 2 μM β-mercaptoethanol, and 5 mM imidazole to remove urea. After removal of urea, the protein was again passing through Ni-NTA affinity beads and eluted with 300 mM imidazole. Sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) and Coomassie staining were used to examine the purity of the protein.
Recombinant Tid1L variants either H121Q [24] or c.452G>C (p.(Arg151Thr)) were generated using a PCR-based method (Table 1). Recombinant nucleotide constructs were verified by DNA sequencing. The Tid1L variants (H121Q and c.452G>C (p.(Arg151Thr)) were purified using the method described previously for wild type Tid1L.
Purification of recombinant Mdj1 variants
Cloning, expression in bacteria and purification of wild type Mdj1 was described previously [24]. An Mdj1 variant c.353G>C (p.(Arg118Thr)), corresponding to the disease-causing variant in the J-domain sequence of Tid1, was generated by PCR-based methods using the primers described in Table 1. The identity of the variant was verified by sequencing. Mdj1 variant were purified using the same method as described above for wild type Mdj1. SDS–PAGE followed by Coomassie staining was used to assess the purity of the protein. The protein absorbance at 280 nm was measured and used to determine its concentration, based on a theoretical extinction co-efficient of 29,380 M−1 cm−1 that was calculated from the amino acid sequence.
Glucose-6-phosphate dehydrogenase (G6PDH) reactivation assay
Refolding activity of Hsp70 variants was examined by monitoring the reactivation of heat aggregated G6PDH, as described previously [24, 25]. Aggregation of G6PDH (0.85 μM) from Leuconostocmesenteroides (Sigma) was carried out in 100 mM Tris (pH 7.4), containing 100 mM KCl, 10 mM MgCl2, and 10 mM DTT for ∼10 min at 52 °C. To measure the refolding of G6DPH, stable aggregates were incubated in ATP regeneration buffer (0.2 M glycine betaine monohydrate, 10 μg/ml pyruvate kinase, 5 mM phosphoenolpyruvate, and 4 mM ATP). After that, the chaperone proteins were added (8 μM of the indicated Hsp70 proteins, 2 or 1 μM of the relevant J-domain protein and 1 μM of the nucleotide exchange factor). The activity was assayed in a solution containing 0.64 µM G6DPH, 3 mM ATP and 0.8 µM NAD+. The increase in absorbance at 340 nm was monitored continuously.
ATPase activity measurement
Steady state ATPase activity of mortalin was followed in a coupled reaction in which phosphoenolpyruvate was converted to pyruvate by pyruvate kinase and the pyruvate was converted to lactate by lactate dehydrogenase [24]. The assay mixture consisted of Hsp70 (8 µM) J-domain protein (2 µM), hMge1 (1 µM), 50 mM Na-HEPES pH 7.4, 50 mM KCl, 10 mM MgCl2, 0.3 mM NADH, 0.2 mM phosphoenolpyruvate, 20 units of pyruvate kinase, and 10 units of lactate dehydrogenase. The reaction was started by the addition of 2 mM ATP. The decrease in the absorbance of NADH was monitored at 340 nm for 4 min. Rates were extracted from the linear phase of the reaction.
Protein purification
Mortalin and hMge1 were purified as described previously [24, 26].
Results
Patient
Patient III-3 (Fig. 1a) is a 15-year-old-female child who was monitored since early childhood due to intellectual disability and a progressively unsteady gait. She was born at term, with a birth weight of 2.4 kg, after an uneventful twin pregnancy and delivery. Her twin brother is healthy. The parents are first cousins and their four other children are normal.

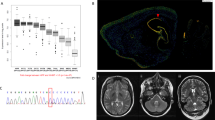
Characterization of the J-domain protein variant in the affected patient. a Family pedigree of the affected patient suffering from developmental delay with intellectual disability and unsteady gait represented by filled circle. The genotype of the variant c.452G>C (p.(Arg151Thr)) in the Tid1 gene is given. b Chromatogram of exon 4 sequence around the variation site. Patient—upper lane, father—middle lane, healthy control—lower lane. c Amino acid sequence alignment of the J domain of Tid1L and closely related homologs. The location of the conserved Arg 151 is indicated by an arrow below the alignment. d The NMR structure of the J domain of Tid1 (2DN9l.pdb) showing the location of the central residues at at the base of the domain. Arg-151 and its interacting residues are labeled. Left panel: wild type. Middle panel: c.452G>C (p.(Arg151Thr)). In the left and middle panels, the ribbon was rendered partially transparent to facilitate visualization of the hydrogen bonds and the labeled residues. Right panel: Expected forces acting on the labeled atoms due to the loss of the positive charge in the variant. Acidic residues are colored red, basic residues are blue, aromatic residues are orange, and polar uncharged residues are yellow. Arrow heads indicate direction of movement that would be expected due to the variation at DNA level. The protein has been rotated in this panel to facilitate visualization of the interacting atoms. Illustrations were generated using the PyMOL program (The PyMOL Molecular Graphics System, version 1.5.0.4; Schrödinger, LLC; available at www.pymol.org).
At infancy, patient III-3 was noted to have a static global developmental delay that progressed during childhood to unsteady gait, regression in intellectual ability and poor school performance. There was no history of seizures, and her hearing was normal. Her ophthalmologic examination showed bilateral moderate hypermetropia (with +4.75 of the right eye and +4.25 of the left eye). The visual evoked potentials were delayed in both eyes indicating optic nerve involvement. In fundus examination, the retina and the optic nerves appear normal.
Physical examination showed a thin child of normal height and head circumference, with no dysmorphism except for pes cavus (Supplementary Fig. S1). Neurological examination revealed an unsteady gait, inability to raise the feet at the level of both ankles (dorsiflexion), mildly decreased control over the distal parts of the upper limbs and significantly decreased control over the distal parts of the lower limbs, with muscle wasting and exaggerated deep-tendon reflexes. Her intelligence was evaluated using the Stanford–Binet Intelligence Scales, 5th edition, with findings of an intelligence quotient (IQ) below the normal values (58) indicating mild to moderate mental retardation. These findings indicate involvement of both the peripheral and central nervous systems.
Laboratory biochemistry tests revealed normal values for metabolic markers including serum ammonia, lactic acid, serum amino acids and urine organic acids.
Brain MRI revealed abnormal bilateral symmetrical signal intensities in the basal ganglia; otherwise normal brain structure was noted including cerebral cortex, ventricular system, cerebellar hemisphere, vermis, and brain stem (Fig. 2).

MRI images of patient’s brain. The images depict bilateral hyper signal (a, b, d) and hypo signal (c) intensity, involving both heads of the caudate nucleus, and the anterior parts of the putamen. e Normal cerebellar brain stem structures
Nerve conduction velocity showed signs of peripheral polyneuropathy. To test for mitochondrial disease, a muscle biopsy was performed, which showed normal enzymatic activities of the five mitochondrial respiratory chain complexes. The patient was diagnosed altogether as having basal ganglia disease of undetermined cause with associated peripheral polyneuropathy, unsteady gait, and intellectual disability.
The disease-causing variation
Whole-exome sequencing resulted in 49.1 million reads with a read depth of X70. Following alignment and variant calling, we removed variants that were called less than X8, were off-target, synonymous, heterozygous, had minor allele frequency (MAF) > 0.5% or were homozygous at GnomAD (http://gnomad.broadinstitute.org/) or MAF > 2% at the Hadassah in-house database (3000 exome analyses). This filtration, together with consideration of evolutionary conservation score (GERP) and additional pathogenicity prediction (SIFT, Mutation Taster), resulted in the identification of a homozygous variant in the DNAJA3 gene HG19 chr16:g.4491398G>C, NM_005147.5: c.452G>C (p.(Arg151Thr)) (Fig. 1b). This amino acid variation is highly conserved among homologs of Tid1 (Fig. 1c) and was confirmed by Sanger sequencing. Segregation analysis showed that the parents and the sisters carry this variant and the brothers are homozygous for the normal allele (Fig. 1b). DNAJA3, also named Tid1, is a J-domain-containing protein that assists mitochondrial Hsp70 (mortalin) in mediating protein folding and disaggregation, but also performs extra-mitochondrial functions. To date, no mitochondrial genetic disease is known to be associated with the folding function of Tid1.
The NMR structure of the J domain of Tid1 (PDB ID: 2DN9) is made of a typical helix-loop-helix (positions 105–147), packed against two smaller helices (positions 94–99 and 148–157) (Fig. 1d). As can be seen, Arg-151 is part of an electrostatic network of residues, which is located at the base of the helix-loop-helix, leading to the stabilization of the packing of the domain. As such, Arg-151, which carries a full positive charge, is stabilized by the negative charges of Asp-155 and Glu-108, the partial negative charge of Gln-106’s side-chain oxygen, the partial charge of Tyr-95’s side-chain oxygen and the π electrons of Tyr-95 (Fig. 1d, left panel). The main chain of Arg-151 is also expected to form hydrogen bonds with the main chains of Asp-147 and Asp-155, both residing on the same helix. Changing Arg-151 into Thr adds a new hydrogen bond between the side chain of Thr-151 and the backbone group of Leu-145 (Fig. 1d, middle panel). However, loss of the guanidinium positive side chain is expected to destabilize the entire electrostatic network described above. In particular, the repulsive interactions between Asp-155, Tyr-95, Tyr-154, and Asp-93 will be more pronounced, without the masking effect of Arg-151 (Fig. 1d, right panel). In summary, loss of the Arg-151 side chain is predicted to significantly destabilize the area of the J domain.
The effect of the disease-causing variant on the import capacity of Tid1
We next examined whether the c.452G>C (p.(Arg151Thr)) variation can affect import of Tid1 precursor, into yeast mitochondria in vitro. As shown in Fig. 3, precursors of both wild type and the c.452G>C (p.(Arg151Thr)) variant were imported into yeast mitochondria. However, the import rate of the c.452G>C (p.(Arg151Thr)) variant was less than that of the wild type protein, leading to less import of the variant (31% and 45%, respectively, after 1 h). As a control, we examined the import of an Su9 (1–69)-DHFR artificial precursor. 20% of this precursor was imported after 1 h.

Import of the Tid1 variant into mitochondria, in vitro. Tid1 wild type or c.452G>C (p.(Arg151Thr)) precursor proteins were labeled with [35S]methionine and imported into mitochondria for 0.5, 20 or 60 min at 30 °C as described. P precursor. M mature protein. -PK sample that was not treated with proteinase K. The samples were subsequently analyzed by SDS–PAGE and autoradiography. A control import system using 1–69 of the yeast DHFR Su9 is provided for comparison. T shows the total amount of precursor that was used for each time point. An asterisk indicates degradation products. A representative experiment is shown
Functional analysis of wild type Tid1 and the p.(Arg151Thr) variant: effect on the co-chaperone function of Tid1
The fact that the Tid1L precursor is efficiently imported into mitochondria rules out an import defect as the sole cause of the disease. We next examined the effect of this c.452G>C (p.(Arg151Thr)) variation on the co-chaperone function of the variant protein. Tid1 and its yeast homolog, called Mdj1, share a high sequence identity (58%) and are interchangeable in their ability to assist Hsp70 in mediating the reactivation of protein aggregates, in vitro [17, 24] and in vivo [27, 28]. One central function of Tid1 is to assist mortalin (human mitochondrial Hsp70) in mediating the reactivation of misfolded or aggregated protein. In this process, Tid1 accelerates ATP hydrolysis to ADP by mortalin, thereby enhancing the binding of unfolded protein to mortalin. This process is reversed by the action of the nucleotide exchange factor (GrpE/Mge1), which accelerates release of the refolded protein. An in vitro reconstitution system of mortalin/Tid1/Mge1 folding activity was developed in our laboratory [24]. We utilized this system in order to examine the effect of the c.452G>C (p.(Arg151Thr)) replacement on the function of Tid1. To this end, we purified wild-type mortalin/Tid1/Mge1 and the Tid1 variant c.452G>C (p.(Arg151Thr)) and examined the effect of the replacement on the function of the team. Finally, we engineered the corresponding replacement in yeast Mdj1 and investigated its effect on the ability of the yeast homolog to function with mortalin.
To examine the effect of the c.452G>C (p.(Arg151Thr)) replacement on the functional interaction of Tid1 with Hsp70 proteins, we first studied the steady state ATPase activity of mortalin in the presence of purified human Mge1 and either wild type yeast Mdj1 or c.353G>C (p.(Arg118Thr)), which contain a replacement corresponding to the disease-causing amino acid in humans. Mortalin alone hydrolyzes ATP with a rate of 0.2 ± 0.025 turnovers/minute (Supplementary Fig S2). In the presence of Mdj1 and Mge1, a 10-fold acceleration in the ATPase activity of mortalin was observed. Under the same conditions, the c.353G>C (p.(Arg118Thr)) variant induced only a 4-fold increase in the ATPase activity of mortalin, indicating a defective interaction between the yeast J-protein and mortalin.
We also examined the effect of the disease-causing variation on ATPase activity in the complete human system. Tid1L, the human J-protein, induced a ~4-fold increase in the ATPase activity of mortalin, in the presence of human Mge1. As a control, we used the known HPD (H121Q) inactive variant, which was less able to stimulate the ATPase activity [24]. Interestingly, the c.452G>C (p.(Arg151Thr)) variant stimulated the ATPase activity of mortalin only ~2-fold, similar to the value obtained with the inactive H121Q variant (Supplementary Fig S2).
In summary, we found that, in vitro, the Tid1 c.452G>C (p.(Arg151Thr)) variant exhibits a reduced ability to function as a J co-chaperone in stimulating ATP hydrolysis by the Hsp70 chaperone, mortalin, suggesting that the replacement causes an impaired interaction between the two proteins.
Since protein remodeling is a central function of the Hsp70 chaperone system inside mitochondria, we next examined whether the compromised ATPase activity of the mortalin/Tid1/Mge1 team correlates with defective disaggregation activity. To this end, we studied the reactivation kinetics of preformed glucose-6-phosphate (G6PDH) aggregates when assisted by Hsp70s in the presence of either wild type or variant [Mdj1 c.353G>C (p.(Arg118Thr))/TidL c.452G>C (p.(Arg151Thr))] proteins. We used a well-established refolding assay whereby thermally denatured, aggregated G6DPH is used as a substrate and spontaneous refolding is quantified by measuring enzyme activity spectrophotometrically as described previously [25]. Native G6DPH was denatured at 52 °C and diluted into folding buffer that contained various combinations of wild type and disease-causing chaperone proteins. Under the conditions of the experiment, the percent of background G6DPH reactivation in the absence of chaperones (∼10%) was used as a control (Fig. 4).

Reactivation of stable G6PDH aggregates by mortalin. a The time dependent activity of G6PDH in the presence of 8 µM mortalin aided by yeast co-chaperone, Mdj1. Mdj1 and Mge1 (red) (2 µM), Mdj1-R118T and Mge1 (1 µM) (green) Mdj1 without Mge1 (blue), Mdj1-R118T (cyan) without Mge1. The activity without any co-chaperones (black). b The yield of G6PDH reactivation obtained, at the end point, by mortalin and indicated yeast co-chaperones. Color code as in a. c The time dependent activity of G6PDH in the presence of 8 µM mortalin aided by human co-chaperones Tid1L (red) (2 µM), Tid1L-R151T (green), Tid1L-H121Q (magenta) all with Mge1 (1 µM). The activity without addition of co-chaperones in buffer as control (black). d The yield of G6PDH reactivation obtained, at the end point, by mortalin and indicated human co-chaperones. The spontaneous refolding of the aggregates in the presence of the reaction buffer is shown (black). Color code as in c
In order to examine the effect of the c.452G>C (p.(Arg151Thr)) changes on the functional interaction of Tid1 with Hsp70 proteins, we first examined the time dependence of reactivation of preformed G6PDH aggregates by mortalin. As presented in Fig. 4, 92% of the aggregated G6PDH was reactivated after 2 h in the presence of wild type Mdj1 and Mge1. Notably, the yield of G6DPH reactivation in the presence of c.353G>C (p.(Arg118Thr)) and Mge1 was lower, reaching only ∼38% of that obtained with wild type protein. The reactivation activity of G6PDH by mortalin in the presence of Mdj1 alone was slightly affected by this DNA variation c.353G>C (p.(Arg118Thr)), reaching ~45% of wild type activity (Fig. 4a, b), compared with ~30% of wild type for c.353G>C (p.(Arg118Thr)), indicating that this change at the DNA level may also be affecting the coordination of the protein with its nucleotide exchange factor, Mge1.
Mortalin is also able to mediate the disaggregation of G6PDH when assisted by Tid1L (Fig. 4c, d) and Mge1, albeit with a lower yield (∼30%). Similarly, to its corresponding yeast co-chaperone variant, human Tid1 c.452G>C (p.(Arg151Thr)) showed compromised co-chaperone function upon assisting mortalin in the presence of hMge1, exhibiting disaggregation activity that is ~50% of that obtained with wild type Tid1L. The level of activity obtained is similar to the activity obtained with the Tid1L-H121Q inactive variant, thereby confirming the specificity of the disaggregation assay used in this study.
Discussion
Numerous neurodegenerative disorders are linked to misfolding and aggregation of proteins. As misfolded, aggregated proteins accumulate, a variety of molecular chaperones is produced to counter protein aggregation and its toxic consequences. Because mammalian mitochondria contain a single Hsp70 (mortalin) and no ClpB-like disaggregating activity has yet been identified in this organelle, mortalin remains the only known mitochondrial chaperone that, together with Tid1, could reactivate protein aggregates. The fact that the mitochondrial Hsp70 chaperone system is induced upon exposure to UPR stress suggests that this system has an important role in mitochondrial proteostasis [29].
In this study, we report the first human mitochondrial disease that is linked to a variant of the human Tid1 protein. The disease is characterized by developmental delay, intellectual disability, unsteady gait and peripheral polyneuropathy. In vitro import assays showed that the c.452G>C (p.(Arg151Thr)) variant is imported into mitochondria in vitro, although at a lower rate than the wild type protein. One explanation for the lower efficiency of the import could be an impaired ability to fully unfold post-translationally-under our assay conditions. Nevertheless, the variant is successfully imported and the lower rate is probably is not sufficient to explain the drastic effects of the c.452G>C (p.(Arg151Thr)) variant that is observed in our patients.
Using an in vitro refolding assay, we examined another possibility, namely the effect of the replacement on the ability of Tid1 to assist Hsp70 in the activation of protein aggregates. Our results clearly show that in the c.452G>C (p.(Arg151Thr)) variant, the chaperone function of the mitochondrial Hsp70 team is severely compromised.
Previous studies have shown that the J-domain of Mdj1/Tid1L is sufficient for regulation of Hsp70 ATPase activity. Based on the NMR structure, it has been suggested that Hsp70 recognizes helix II-HPD and that this is the minimal unit of the J-domain that is recognized by Hsp70. Consistent with this proposal, synthetic peptides that correspond to helix II and the HPD tripeptide were recognized by Hsp70. However, the J-domain peptide was one-tenth as efficient as the actual J-domain of Hsp40 at blocking interactions between Hsp40 and Hsp70 [30]. Perhaps the helix II-HPD sequence is the minimal sequence recognized by Hsp70, but the presence of adjacent regions in the J-domain, such as helix III and helix-IV are required to stabilize this structure in a high affinity conformation.
References
Bukau B, Horwich AL. The Hsp70 and Hsp60 chaperone machines. Cell. 1998;92:351–66.
Bukau B, Weissman J, Horwich A. Molecular chaperones and protein quality control. Cell. 2006;125:443–51.
Hartl FU, Bracher A, Hayer-Hartl M. Molecular chaperones in protein folding and proteostasis. Nature. 2011;475:324–32.
Gething M-J, Sambrook J. Protein folding in the cell. Nature. 1992;355:33–45.
Craig E, Weissman J, Horwich A. Heat shock proteins and molecular chaperones: mediators of protein conformation and turnover in the cell. Cell. 1994;78:365–72.
Hartl FU, Hayer-Hartl M. Protein folding. Molecular chaperones in the cytosol: from nascent chain to folded protein. Science. 2002;295:1852–8.
Bhattacharyya T, Karnezis AN, Murphy SP, Hoang T, Freeman BC, Phillip B et al. Cloning and subcellular localization of human mitochondrial hsp70. J Biol Chem. 1995;270:1705–10.
Syken J, De-Medina T, Münger K. TID1, a human homolog of the Drosophila tumor suppressor l(2)tid, encodes two mitochondrial modulators of apoptosis with opposing functions. Proc Natl Acad Sci USA. 1999;96:8499–504.
Craig EA, Kramer J, Kosic-Smithers J. SSC1, a member of the 70-kDa heat shock protein multigene family of Saccharomyces cerevisiae, is essential for growth. Proc Natl Acad Sci USA. 1987;84:4156–60.
Voos W. Chaperone-protease networks in mitochondrial protein homeostasis. Biochim Biophys Acta. 2013;1833:388–99.
Karzai AW, McMacken R. A bipartite signaling mechanism involved in DnaJ-mediated activation of the Escherichia coli DnaK protein. J Biol Chem. 1996;271:11236–46.
Liberek K, Marszalek J, Ang D, Georgopoulos C, Zylicz M. Escherichia coli DnaJ and GrpE heat shock proteins jointly stimulate ATPase activity of DnaK. Proc Natl Acad Sci. 1991;88:2874–8.
Brehmer D, Rüdiger S, Gässler CS, Klostermeier D, Packschies L, Reinstein J et al. Tuning of chaperone activity of Hsp70 proteins by modulation of nucleotide exchange. Nat Struct Biol. 2001;8:427–32.
Shomura Y, Dragovic Z, Chang H-C, Tzvetkov N, Young JC, Brodsky JL et al. Regulation of Hsp70 Function by HspBP1. Mol Cell. 2005;17:367–79.
Wadhwa R, Kaul SC, Ikawa Y, Sugimoto Y. Identification of a novel member of mouse hsp70 family. Its association with cellular mortal phenotype. J Biol Chem. 1993;268:6615–21.
Wadhwa R, Sugihara T, Yoshida A, Nomura H, Raddel RR, Simpson R et al. Selective toxicity of MKT-077 to cancer cells is mediated by its binding to the hsp70 family protein mot-2 and reactivation of p53 function. Cancer Res. 2000;60:6818–21.
Goswami AV, Chittoor B, D’Silva P. Understanding the functional interplay between mammalian mitochondrial Hsp70 chaperone machine components. J Biol Chem. 2010;285:19472–82.
Iosefson O, Azem A. Reconstitution of the mitochondrial Hsp70 (mortalin)-p53 interaction using purified proteins—identification of additional interacting regions. FEBS Lett. 2010;584:1080–4.
Kaul SC, Deocaris CC, Wadhwa R. Three faces of mortalin: a housekeeper, guardian and killer. Exp Gerontol. 2007;42:263–74.
Jin J, Hulette C, Wang Y, Zhang T, Pan C, Wadhwa R et al. Proteomic identification of a stress protein, mortalin/mthsp70/GRP75: relevance to Parkinson disease. Mol Cell Proteom. 2006;5:1193–204.
Mihara M, Erster S, Zaika A, Petrenko O, Chittenden T, Pancoska P et al. p53 has a direct apoptogenic role at the mitochondria. Mol Cell. 2003;11:577–90.
Schuler M, Green DR. Mechanisms of p53-dependent apoptosis. Biochem Soc Trans. 2001;29:684–8.
Mokranjac D, Paschen SA, Kozany C, Prokisch H, Hoppins SC, Nargang FE et al. Tim50, a novel component of the TIM23 preprotein translocase of mitochondria. EMBO J. 2003;22:816–25.
Iosefson O, Sharon S, Goloubinoff P, Azem A. Reactivation of protein aggregates by mortalin and Tid1-the human mitochondrial Hsp70 chaperone system. Cell Stress Chaperon. 2012;17:57–66.
Diamant S, Peres Ben-Zvi A, Bukau B, Goloubinoff P. Size-dependent disaggregation of stable protein aggregates by the DnaK chaperone machinery. J Biol Chem. 2000;275:21107–13.
Horst M, Oppliger W, Rospert S, Schönfeld HJ, Schatz G, Azem A. Sequential action of two hsp70 complexes during protein import into mitochondria. EMBO J. 1997;16:1842–9.
Lu B, Garrido N, Spelbrink JN, Suzuki CK. Tid1 isoforms are mitochondrial DnaJ-like chaperones with unique carboxyl termini that determine cytosolic fate. J Biol Chem. 2006;281:13150–8.
Rowley N, Prip-Buus C, Westermann B, Brown C, Schwarz E, Barrell B et al. Mdj1p, a novel chaperone of the DnaJ family, is involved in mitochondrial biogenesis and protein folding. Cell. 1994;77:249–59.
Zhao Q, Wang J, Levichkin IV, Stasinopoulos S, Ryan MT, Hoogenraad NJ. A mitochondrial specific stress response in mammalian cells. EMBO J. 2002;21:4411–9.
Tsai J, Douglas MG. A conserved HPD sequence of the J-domain is necessary for YDJ1 stimulation of Hsp70 ATPase activity at a site distinct from substrate binding. J Biol Chem. 1996;271:9347–54.
Acknowledgements
This work was supported by the DFG trilateral project (Reference number SCHO 754/5-2) and the Israel Science Foundation ISF-1507/13.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Patra, M., Weiss, C., Abu-Libdeh, B. et al. A novel variant of the human mitochondrial DnaJ protein, Tid1, associates with a human disease exhibiting developmental delay and polyneuropathy. Eur J Hum Genet 27, 1072–1080 (2019). https://doi.org/10.1038/s41431-019-0358-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-019-0358-9
This article is cited by
-
Putting human Tid-1 in context: an insight into its role in the cell and in different disease states
Cell Communication and Signaling (2022)
-
Mitochondrial “dysmorphology” in variant classification
Human Genetics (2022)
