
Abstract
Silver-Russell syndrome is an imprinting disorder characterized by severe intrauterine and postnatal growth retardation. The majority of patients show loss of methylation (LOM) of the H19/IGF2 IG-DMR (ICR1) in 11p15.5. In ~10% of these patients aberrant methylation of additional imprinted loci on other chromosomes than 11 can be observed (multilocus imprinting defect – MLID). Recently, genomic variations in the ICR1 have been associated with disturbed methylation of the ICR1. In addition, variants in factors contributing to the life cycle of imprinting are discussed to cause aberrant imprinting, including MLID. These variants can either be identified in the patients with imprinting disorders themselves or in their mothers. We performed comprehensive studies to elucidate the role of both cis-acting variants in 11p15.5 as well as of maternal effect variants in the etiology of ICR1 LOM. Whereas copy number analysis and next generation sequencing in the ICR1 did not provide any evidence for a variant, search for maternal effect variants in 21 mothers of patients with ICR1 LOM identified two carriers of NLRP5 variants. By considering our results as well as those from the literature, we conclude that the causes for epimutations are heterogeneous. MLID might be regarded as an own etiological subgroup, associated with maternal effect variants in NLRP and functionally related genes. In addition, these variants might also contribute to LOM of single imprinted loci. Furthermore, genomic variants in the patients themselves might result in aberrant methylation patterns and need further investigation.
Similar content being viewed by others

Introduction
Silver–Russell syndrome (SRS, OMIM 180860) is a congenital imprinting disorder, characterized by severe intrauterine and postnatal growth retardation, relative macrocephaly, a protruding forehead, body asymmetry, feeding difficulties and further less frequent features (for review: ref. [1]). In 30–60% of patients molecular disturbances of the imprinting control region 1 (ICR1) in 11p15.5 can be observed, consisting of loss of methylation (LOM) of the H19/IGF2 IG-DMR (differentially methylated region) in the majority of cases, and copy number variations (duplications, deletions). In peripheral lymphocytes of nearly 10% of SRS patients with ICR1 LOM, aberrant methylation of additional imprinted loci on other chromosomes than 11 can be observed (so-called multilocus imprinting disturbance – MLID [2]). However, MLID commonly occurs in mosaic state, and by testing additional tissues the number of positive MLID cases in the ICR1 LOM cohort increases up to 38% [3]. MLID is also detectable in other imprinting disorders, in particular in the overgrowth disorder Beckwith–Wiedemann syndrome (BWS; OMIM 130650) where it accounts for more than 12% of cases.
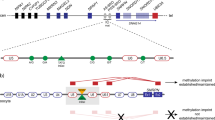
The ICR1 regulates the fine-tuned expression of two imprinted genes in 11p15.5, the paternally expressed growth factor IGF2, and the maternally expressed H19 gene (for review: ref. [4]). It includes several binding and recognition sites for methylation-specific DNA binding factors (Fig. 1). The maternal ICR1 allele is unmethylated and enables the binding of the CCCTC-binding factor (CTCF). The binding of CTCF is required to maintain the unmethylated status of the allele and to inhibit the interaction of the IGF2 promoters with downstream enhancer motifs that are shared between IGF2 and H19 (for review: ref. [5]). Vice versa, the paternal ICR1 copy is methylated, thereby preventing the binding of CTCF and allowing the expression of IGF2. Among other factors, the KRAB zinc finger protein ZFP57 [6] mediates the maintenance of the paternal ICR1 methylation marks but also of other paternal as well as maternal imprints during embryonic epigenetic reprogramming. Both CTCF and ZFP57 binding sites cluster in the repetitive modules of the ICR1 [7]. Genomic variants in ZFP57 have previously been identified to cause hypomethylation of specific genes, associated with the imprinting disorder TNDM (transient neonatal diabetes mellitus) [8]. However, variants affecting the function of ZFP57 have been excluded to contribute to both ICR1 LOM in SRS or imprinting control region 2 (ICR2) LOM in BWS [9, 10]. In addition, the ICR1 also harbors binding sites for the pluripotency factors OCT4 and SOX2. Recently, genomic variations in the ICR1 have been associated with LOM in SRS and gain of methylation (GOM) in BWS [11, 12]. Additionally, copy number variations of different extent spanning several of the binding sites and repetitive modules of the ICR1 can be observed in SRS and BWS patients, but also in healthy individuals (for review: ref. [13]), which therefore allow further delineation of functional elements in 11p15.5.

Structure of the ICR1 in 11p15.5 containing A and B-type repeats as well as CTCF and ZFP57 binding sites (yellow lollipops) and OCT4 binding sites (red pentagon) based on reports from the literature and predicted consensus sequences (hg19). ZFP57 binding sites on the reverse strand are not shown (for detailed information see ref. [7]). The whole region with its binding sites was covered by the 11p15.5-NGS assay
In addition to these cis-acting elements in 11p15.5, variants in factors contributing to the establishment, maintenance and erasure of imprinting marks during development have been identified to cause aberrant imprinting. These variants can be detected either in the patients themselves (e.g. in the aforementioned gene ZFP57 [8]) or in their mothers (for review: ref. [14]). In particular, the primordial germ cells of a developing embryo as well as the (early) embryo itself undergo an epigenetic reprogramming. In the early embryo, maternally-provided factors in the ooplasm are required for the maintenance of the imprinting marks. In case the mother carries variants in one of the genes encoding these factors, aberrant imprints occur in their offspring (so-called maternal-effect variant). In fact, there is a growing number of reports corroborating this observation (for review: ref. [14]). Several of these genes are members of the NLRP gene family, among them NLRP2, NLRP5, and NLRP7. Maternal-effect variants in all three genes have been postulated to be associated with reproductive problems, including pregnancy losses, infertility, and MLID in liveborn children. As mentioned before, MLID is present in a significant number of patients with SRS and BWS, and in fact children born to mothers with NLPR2, NLRP5, and NLRP7 maternal effect variants often exhibit features of these imprinting disorders [15,16,17,18].
In addition to genomic factors modifying the imprinting patterns in an individual, also environmental factors are discussed to affect the embryonic reprogramming. Since several years it is known that ART technologies can affect the methylation status of some genes in mammals (e.g. refs. [19, 20]), and it has been suggested that the establishment and maintenance of DNA methylation of imprinted regions might be disturbed by the use of fertility drugs as well as by in-vitro culture of embryos. On the other hand, infertility itself might be regarded as a risk factor for congenital anomalies, including imprinting disorders (e.g. ref. [21]).
Based on these observations, we performed comprehensive studies to elucidate the role of both cis-acting variants in 11p15.5 as well as of maternal effect variants in the etiology of ICR1 LOM in a cohort of SRS patients.
Patients
The total study cohort consisted of 83 patients ascertained for routine molecular diagnostics of SRS, some of these patients have been reported in previous studies (e.g. refs. [17, 18, 22]). In case clinical data were provided, clinical scoring for SRS was performed by applying the Netchine-Harbison (NH) scoring system [1]. The study was approved by the Ethical Review Board of the University Hospital Aachen (Germany). In all patients, DNA was extracted from peripheral lymphocytes. In 21 families, genomic DNA samples from the mothers were available.
Materials and Methods
Methylation-specific assays
All samples (n = 83) were screened for 11p15 copy number variations (CNVs) and aberrant methylation of the imprinted loci in 11p15 (H19/IGF2 IG-DMR, KCNQ1OT1 TSS DMR) and two loci on chromosome 7 (GRB10 alt-TSS DMR (chr7p12); MEST alt-TSS DMR (chr 7q32)) by methylation-specific multiplex ligation-dependent probe amplification (MS-MLPA) assays (kits ME030, ME032; MRC Holland, Amsterdam, NL). Testing for further imprinted loci associated with imprinting disorders was performed either by MS MLPA (ME032, ME034; MRC Holland) or by a methylation-specific single nucleotide primer extension (MS-SNuPE) assay [23]. The loci examined included: PLAGL1 TSS alt-DMR (chr6q24), IGF2R Int2 DMR (chr6q25), MEG3 TSS DMR (chr14q32), SNURF TSS DMR (chr15q11.2), GNAS-AS1 TSS DMR (chr20q13.32), and GNAS A/B TSS DMR (chr20q13.32).
CNV analysis
For the detection of further genomic imbalances in ICR1 LOM patients (n = 34) either the Affymetrix GeneChip®Genome-Wide Human SNP 6.0-Array or the CytoScan® HD Array (Affymetrix, Santa Clara/CA, USA) were used. In general, CNVs > 200 kb with a mean marker distance of < 5 kb were considered, but the ICR1 and ICR2 regions in 11p15 were analyzed on single SNP or probe basis.
NGS analyses
To determine cis-acting SNVs in 11p15.5 in ICR1 LOM carriers (n = 23), a targeted NGS panel (Nextera rapid capture custom enrichment) was designed using the Illumina Design Studio (Illumina, San Diego/CA, USA). The panel comprised the CTCF/ZFP57 and OCT4/SOX2 binding sites (Fig. 1) and spanned the whole ICR1 and ICR2 regions (hg 19: 1,742,917–2,911,076).
For the analysis of the mothers of ICR1 LOM carriers (n = 21), the custom enrichment included the NLRP genes and two additional genes suggested as candidate genes for MLID and setting of imprinting marks (NLRP2–14, TRIM28, and KHDC3L). This assay has been reported previously [24] (for the used transcripts see suppl. Table 1).
Library preparation was carried out following the manufacturer’s protocol. DNA fragmentation was achieved by enzymatic tagmentation. The libraries were sequenced on a MiSeq benchtop Sequencer with 2 × 151 cycles and the v2 reagent kit using paired end reads. Primary data (fast q) was aligned to the hg19 reference genome and then processed by the integrated MiSeq Reporter software to generate vcf (variant call format) files. Variant annotation was carried out using the Illumina Variant Studio (version 2.2.3). For the detection of maternal effect variant, the following filters were applied to reduce the number of detected variants. In a first step variants with a minor allele frequency (MAF) >1% were excluded from analysis [1000 Genomes project database (April 2012 phase 1 call set) or in the Exome Variant Server (NHLBI GO Exome Sequencing Project (ESP), Seattle, WA (evs.gs.washington.edu/EVS) [November 2012 accessed)]. In a second step synonymous variants were excluded.
For the chromosome 11p15.5 region NGS assay the averaged target region coverage (20×) was >90%, for the maternal effect variant panel it was >97% (20×).
Results
By routine diagnostic testing for SRS, 83 patients were identified to carry an ICR1 LOM (suppl. Table 2). In 46 patients, sufficient clinical data were available to apply the clinical NH score: in 40 patients the clinical diagnosis could be confirmed because they showed at least 4 out of 6 items included in the NH score, whereas in six patients <3 of the six features were present. Data on the mode of conception were available in 34 families: seven patients were born after assisted reproduction (IvF, ICSI)(~20.6%).
Testing for further imprinted loci revealed a MLID in 10.4% (n = 9). As reported previously, search for maternal effect variants identified putative variants affecting the function of NLRP2, NLRP5, and PADI6 in mothers of three of these patients ([17, 18]; supplementary table 2). In eight of these patients the clinical data indicated a SRS (≥4 NH score items), in another patient only two features were present.
MS-MLPA analysis did not show any copy number variation (CNV) at the imprinted loci tested. By genomewide SNP array analysis in 35 patients, no clinically relevant deletions or duplications could be detected, neither in 11p15.5 nor in other chromosomal regions.
The targeted NGS approach aiming to identify SNVs in 11p15.5 in 23 patients did not reveal any evidence for functionally relevant genomic variants in the OCT4/SOX2 and CTCF/ZFP57 binding sites.
In the 21 mothers of patients with isolated ICR1 LOM, we identified two rare and possibly variants affecting the function of NLRP5 (9.5%) (Table 1) but neither in NLRP2 and NLRP7 nor in one of the other targeted genes we identified any variant of putative pathogenic relevance.
In the mother of patient SRS-35, heterozygosity for the NLRP5 variant NM_153447.4:c.68 T > A (p.(Val23Asp); rs753824534) was identified, it was also present in the child. The patient was the first child of healthy unrelated parents, a family history of reproductive failure and abortions was not reported. Clinically, the patient showed 4 out of 6 features from the NH score, he did not exhibit relative macrocephaly and feeding difficulties.
Heterozygosity for the NLRP5 variant NM_153447.4:c.3259 G > A (p.(Glu1087Lys), rs762535392) was detected in the mother of patient SRS38, it was not inherited to the patient. This patient showed all NH scoring items. Family history was unremarkable, too.
In fact, both variants have been listed in the databases but they are rare in control cohorts with very low MAFs (minor allele frequency). Their occurrence in control datasets is compatible with their nature as maternal-effect variant, as they are only identified as functionally relevant in women during pregnancy. The pathogenicity prediction for the two variants is differing (Table 1). The variant c.68 T > A (p.(Val23Asp) is predicted as deleterious or pathogenic by two variant effect prediction programs, whereas mutation taster indicates that it is an apathogenic variant. It does not affect any of the known functional domains but a functional significance cannot be excluded. In contrast to the rather pathogenic nature of p.(Val23Asp), the variant c.3259 G > A (p.(Glu1087Lys) is categorized as benign by all applied prediction tools. It lies within the 12th leucine rich repeat (LRR) of NLRP5 which are postulated to function as ligand sensors of NLRP proteins [25].
Discussion
Though the association of ICR1 LOM with SRS has been established more than 10 years ago [26], its underlying cause is still unknown. However, with the increasing identification of cis-acting elements in 11p15.5 influencing the imprinting status of the ICR1 [13, 27, 28] and of maternal-effect variants probably affecting the maintenance of imprints in the oocyte (for review: ref. [29]), there is growing evidence that a significant number of cases with ICR1 LOM in SRS and GOM in BWS can be regarded as secondary epimutations caused primarily by genomic variants, either in the patient or his/her mother. By analyzing a cohort of ICR1 LOM carriers and their mothers for cis-acting factors and maternal effect variants, we aimed to estimate the contribution of these factors to the etiology of imprinting mark alterations in a defined patient cohort.
In a first step, the frequency of MLID carriers in our cohort was determined. By testing 10 imprinted loci on six chromosomes in lymphocyte DNA from 83 patients ascertained for SRS routine testing, we identified nine ICR1 LOM carriers with additional altered imprinting marks. This MLID frequency of ~10% corresponds to that from other studies in the literature (for review: ref. [22]). However, this frequency is based on results from lymphocyte DNA analyses. As Azzi et al. [30] recently showed by analyses of other tissues than lymphocytes, the frequency of MLID among SRS is probably higher due to the mosaic distribution of the epimutation: In up to 38% of patients the group identified altered imprinting patterns compatible with MLID. Thus, it can be assumed that a significant number of SRS patients currently diagnosed as “isolated” ICR1 LOM carriers are MLID patients. Clinically, MLID carriers often exhibit the common SRS phenotype, though additional features uncommon for SRS can be present (e.g. refs. [13, 31]). In summary, it can be questioned whether patients with MLID and those with apparently isolated ICR1 LOM share the same pathoetiology, or whether the aberrant methylation patterns are caused by different mechanism.
To answer this question, we searched for molecular alterations in genes contributing to the subcortical maternal complex as protein complex essential for zygote progression in the early embryo. For MLID families it could be shown that maternal effect variants in these factors (i.e. NLRP2, NLRP5, and NLRP7) can be identified in more than 50% (20 out of 38 cases [17, 18]). However, for families with isolated ICR1 LOM these genes have not yet been investigated. By analyzing 21 mothers of ICR1 LOM carriers, we identified two variants in the NLRP5 gene. In fact, the bioinformatic predictions for these variants do not convincingly proof evidence for their pathogeneity, and these variants are listed as very rare in the databases. However, if a functional role as maternal effect variants is considered, it is well conceivable that they occur in control populations because they become functional only in the offspring of female carriers, not in the proband himself/herself, and therefore escape the identification as pathogenic. Additionally, the average target region coverage (20×) did not allow the identification of mosaicism of putative pathogenic variants.
The identification of putative variants in the NLRP5 gene is compatible with data from other studies: in offspring of MLID mothers with NLRP5 variants both maternally and paternally methylated loci are hypomethylated [17], suggesting the role of NLRP5 in the post-zygotic maintenance of imprints.
Interestingly, a similar role has been suggested for NLRP2 based on observations in Nlrp2-/- knock-out mice [32], and the first NLRP2 variant in a MLID family with a SRS phenotype has been reported recently [18]. However, variants in NLRP2 in MLID/SRS families seem to be less frequent, and among the mothers of ICR1 LOM carriers NLRP2 variants were not detectable. On the other hand, the lack of variants in NLRP7 in the cohort of families with the loss of the paternal imprint at the ICR1 is compatible with the observation that NRLP7 variants are exclusively associated with the loss of maternal imprints, corresponding to its suggested role in oocyte-specific methylation establishment (for review: ref. [14]).
In fact, both mothers carrying the NLRP5 variants were heterozygous, and our NGS studies did not provide any clue to a second variant in the coding regions or exon/intron boundaries. Though the majority of NLRP5 variant carriers reported so far show homozygosity or compound-heterozygosity, heterozygosity for a single variant associated with MLID has already been reported [17], and from observations in carriers of variants in NLRP7 and other maternal effect genes there is growing evidence for an autosomal dominant maternal effect of the variants [15, 18]. It can be speculated that heterozygosity for NLRP5 variants contributes to a milder phenotype, in the sense that only single imprinted loci are affected and that reproductive failure is not observed.
In summary, we hypothesize that apparent isolated ICR1 LOM is the result of heterozygous maternal effect variants in NLRP5. In fact, in lymphocyte DNA of the patients the ICR1 LOM was the only locus out of 10 imprinted regions with aberrant methylation, but further untested loci might be affected as well. Furthermore, a mosaic distribution of MLID might delude an isolated ICR1 LOM in lymphocytes in the patients from our NLRP5 variant families. Unfortunately, we could not determine whereas in other tissues MLID was present. The latter is corroborated by the data from Azzi et al. [3] showing that the number of MLIDs among ICR1 LOM cases increases up to 38% when additional tissues are analyzed.
Therefore further studies are needed to clarify whether NLRP variants are restricted to MLID or whether they also account for imprinting defects of single loci. However, it can also be hypothesized that heterozygous NLRP5 variants resemble a milder molecular phenotype than homozygous variants leading to MLIDs.
Another molecular change resulting in altered ICR1 imprinting patterns are single nucleotide variants (SNVs) and copy number variants (CNVs) in the OCT4/SOX2 binding sites or neighbored sequences in single SRS and BWS patients with ICR1 LOM or GOM, respectively [7, 11, 28]. We therefore aimed to identify SNVs and CNVs in the regulative elements close to the ICR1 by a targeted NGS approach, MLPA and SNP array typing but we could not identify any relevant sequence variant. Thus, we did not get any evidence for a significant role of molecular changes in the ICR1, neither of deletions or duplications in 83 ICR1 LOM patients nor of SNVs in 23 patients investigated by NGS. In fact, we cannot exclude that SNVs are responsible for the epimutation in single patients, as we analyzed only a subcohort of patients by NGS, furthermore smaller CNVs or CNVs with other extents not covered by the MLPA and array approaches also escape the detection. However, we could not detect CNVs corresponding to those reported by Al Habib and coworkers [28] which were detected with the same MLPA approach used in this study. In fact, Al Habib et al. identified only three CNVs in a cohort of 234 ICR LOM patients (~1.3%), thus it is conceivable that we missed a similar variant due to the smaller size of our population (n = 83).
However, it is obvious that CNVs and SNVs in the ICR1 region do not significantly contribute to the etiology of ICR1 LOM in SRS. Nevertheless, CNV tests should be included in the diagnostic testing strategy [33], though they represent a rare cause of this epimutation (<1% (3/317 cases). SNVs in the ICR1 have been identified to cause GOM in BWS, suggesting that OCT4/SOX2 binding protects against methylation of this region [28]. Due to this function, SNVs in OCT4/SOX2 as well as CTCF binding sites probably do not cause hypomethylation of the paternal ICR1 allele, but it can be hypothesized that alterations of the ZFP57 binding sites disturb the maintenance of the paternal ICR1 imprinting marks. The lack of genomic variants affecting this binding site is consistent with the exclusion of variants in ZFP57 in ICR1 LOM patients [10]. As ZFP57 variants cause the PLAGL1-DMR
hypomethylation in TNDM [8], it can rather be postulated that the ZFP57 mediated regulation of imprinting marks is locus-specific, and only of minor importance if at all for the regulation of the ICR1 in 11p15.5.
Conclusion
Although aberrant methylation patterns at imprinted loci significantly contribute to the spectrum of molecular alterations in Imprinting Disorders, the knowledge on the molecular causes for the aberrant imprints is limited. Based on a systematic screening for causative factors in a representative cohort of patients with apparently the same epimutation, we tried to contribute to the understanding of the life cycle of imprinting. We confirm that the causes for altered imprinting patterns at least in ICR1 LOM carriers are heterogeneous for the following reasons. (a) The number of MLID carriers among patients with at first glance single epimutations might be larger but they currently escape detection due to mosaic distribution of the MLID. Further methylome wide studies as well as the analysis of further tissues might help to confirm or discard the presence of isolated ICR1 LOM. In fact, the MLID subcohort might be regarded as an own etiological subgroup, associated with maternal effect variants in NLRP and related genes. However, NLRP5 variants might contribute to the etiology of ICR1 LOM and SRS. (b) In some cases, exogenic factors like ART might cause altered imprinting patterns. (c) Genomic variants in the patients themselves might result in aberrant methylation patterns and need further investigation.
References
Wakeling EL, Brioude F, Lokulo-Sodipe O, O'Connell SM, Salem J, Bliek J. Diagnosis and management of Silver-Russell syndrome: first international consensus statement. Nat Rev Endocrinol. 2017;13:105–24.
Mackay DJ, Eggermann T, Buiting K, et al. Multilocus methylation defects in imprinting disorders. Biomol Concepts. 2015;6:47–57.
Azzi S, Blaise A, Steunou V, et al. Complex tissue-specific epigenotypes in Russell-Silver Syndrome associated with 11p15 ICR1 hypomethylation. Hum Mutat. 2014;35:1211–20.
Azzi S, Abi Habib W, Netchine I. Beckwith-Wiedemann and Russell-Silver Syndromes: from new molecular insights to the comprehension of imprinting regulation. Curr Opin Endocrinol Diabetes Obes. 2014;21:30–8.
Hur SK, Freschi A, Ideraabdullah F, et al. Humanized H19/Igf2 locus reveals diverged imprinting mechanism between mouse and human and reflects Silver-Russell syndrome phenotypes. Proc Natl Acad Sci USA. 2016;113:10938–43.
Li X, Ito M, Zhou F, Youngson N, et al. A maternal-zygotic effect gene, Zfp57, maintains both maternal and paternal imprints. Dev Cell. 2008;15:547–57.
Sparago A, Cerrato F, Riccio A. Is ZFP57 binding to H19/IGF2:IG-DMR affected in Silver-Russell syndrome? Clin Epigenetics. 2018;10:23.
Mackay DJ, Callaway JL, Marks SM, et al. Hypomethylation of multiple imprinted loci in individuals with transient neonatal diabetes is associated with mutations in ZFP57. Nat Genet. 2008;40:949–51.
Boonen SE, Hahnemann JM, Mackay D, et al. No evidence for pathogenic variants or maternal effect of ZFP57 as the cause of Beckwith-Wiedemann Syndrome. Eur J Hum Genet. 2012;20:119–21.
Spengler S, Gogiel M, Schonherr N, Binder G, Eggermann T. Screening for genomic variants in ZFP57 in Silver-Russell syndrome patients with 11p15 epimutations. Eur J Med Genet. 2009;52:415–6.
Abi Habib W, Azzi S, Brioude F, et al. Extensive investigation of the IGF2/H19 imprinting control region reveals novel OCT4/SOX2 binding site defects associated with specific methylation patterns in Beckwith-Wiedemann syndrome. Hum Mol Genet. 2014;23:5763–73.
Demars J, Rossignol S, Netchine I, et al. New insights into the pathogenesis of Beckwith-Wiedemann and Silver-Russell syndromes: contribution of small copy number variations to 11p15 imprinting defects. Hum Mutat. 2011;32:1171–82.
Begemann M, Spengler S, Gogiel M, et al. Clinical significance of copy number variations in the 11p15.5 imprinting control regions: new cases and review of the literature. J Med Genet. 2012;49:547–53.
Monk D, Sanchez-Delgado M, Fisher R. NLRPs, the subcortical maternal complex and genomic imprinting. Reproduction. 2017;154:R161–R70.
Soellner L, Begemann M, Degenhardt F, Geipel A, Eggermann T, Mangold E. Maternal heterozygous NLRP7 variant results in recurrent reproductive failure and imprinting disturbances in the offspring. Eur J Hum Genet. 2017;25:924–9.
Meyer E, Lim D, Pasha S, et al. Germline mutation in NLRP2 (NALP2) in a familial imprinting disorder (Beckwith-Wiedemann Syndrome). PLoS Genet. 2009;5:e1000423.
Docherty LE, Rezwan FI, Poole RL, et al. Mutations in NLRP5 are associated with reproductive wastage and multilocus imprinting disorders in humans. Nat Commun. 2015;6:8086.
Begemann M, Rezwan FI, Beygo J, et al. Maternal variants in NLRP and other maternal-effect proteins associated with multi-locus imprinting disturbance in offspring. J Med Genet. 2018;55:497–504. in press
Chen Z, Hagen DE, Elsik CG, et al. Characterization of global loss of imprinting in fetal overgrowth syndrome induced by assisted reproduction. Proc Natl Acad Sci USA. 2015;112:4618–23.
de Waal E, Yamazaki Y, Ingale P, et al. Primary epimutations introduced during intracytoplasmic sperm injection (ICSI) are corrected by germline-specific epigenetic reprogramming. Proc Natl Acad Sci USA. 2012;109:4163–8.
Buckett WM, Tan SL. Congenital abnormalities in children born after assisted reproductive techniques: how much is associated with the presence of infertility and how much with its treatment? Fertil Steril. 2005;84:1318–9. discussion 27
Eggermann T, Heilsberg AK, Bens S, et al. Additional molecular findings in 11p15-associated imprinting disorders: an urgent need for multi-locus testing. J Mol Med. 2014;92:769–77.
Begemann M, Leisten I, Soellner L, et al. Use of multilocus methylation-specific single nucleotide primer extension (MS-SNuPE) technology in diagnostic testing for human imprinted loci. Epigenetics. 2012;7:473–81.
Soellner L, Kopp KM, Mutze S, et al. NLRP genes and their role in preeclampsia and multi-locus imprinting disorders. J Perinat Med. 2017;46:169–73.
Proell M, Riedl SJ, Fritz JH, Rojas AM, Schwarzenbacher R. The Nod-like receptor (NLR) family: a tale of similarities and differences. PLoS One. 2008;3:e2119.
Gicquel C, Rossignol S, Cabrol S, et al. Epimutation of the telomeric imprinting center region on chromosome 11p15 in Silver-Russell syndrome. Nat Genet. 2005;37:1003–7.
Berland S, Appelback M, Bruland O, et al. Evidence for anticipation in Beckwith-Wiedemann syndrome. Eur J Hum Genet. 2013;21:1344–8.
Abi Habib W, Brioude F, Azzi S, et al. 11p15 ICR1 partial deletions associated with IGF2/H19 DMR hypomethylation and Silver-Russell syndrome. Hum Mutat. 2017;38:105–11.
Sanchez-Delgado M, Martin-Trujillo A, Tayama C, et al. Absence of maternal methylation in biparental hydatidiform moles from women with NLRP7 maternal-effect mutations reveals widespread placenta-specific imprinting. PLoS Genet. 2015;11:e1005644.
Azzi S, Steunou V, Tost J, et al. Exhaustive methylation analysis revealed uneven profiles of methylation at IGF2/ICR1/H19 11p15 loci in Russell Silver syndrome. J Med Genet. 2015;52:53–60.
Bliek J, Terhal P, van den Bogaard MJ, et al. Hypomethylation of the H19 gene causes not only Silver-Russell syndrome (SRS) but also isolated asymmetry or an SRS-like phenotype. Am J Hum Genet. 2006;78:604–14.
Mahadevan S, Sathappan V, Utama B, et al. Maternally expressed NLRP2 links the subcortical maternal complex (SCMC) to fertility, embryogenesis and epigenetic reprogramming. Sci Rep. 2017;7:44667.
Eggermann K, Bliek J, Brioude F, et al. EMQN best practice guidelines for the molecular genetic testing and reporting of chromosome 11p15 imprinting disorders: Silver-Russell and Beckwith-Wiedemann syndrome. Eur J Hum Genet. 2016;24:1377–87.
Acknowledgements
This work was funded by the Bundesministerium für Bildung und Forschung (BMBF: 01GM1513B) and the Deutsche Forschungsgemeinschaft (DFG, EG110/15–1; 948/32–1 FUGG). This work was supported by the Genomics Facility, a core facility of the Interdisciplinary Center for Clinical Research (IZKF) Aachen within the Faculty of Medicine at RWTH Aachen University.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Online databases and prediction tools: dbSNPhttps://www.ncbi.nlm.nih.gov/projects/SNP/; gnomADhttp://gnomad.broadinstitute.org/; LOVD https://databases.lovd.nl/shared/genes/NLRP5; MutationTasterhttp://www.mutationtaster.org/; PolyPhen2http://genetics.bwh.harvard.edu/pph2/; SIFThttp://sift.jcvi.org/
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Soellner, L., Kraft, F., Sauer, S. et al. Search for cis-acting factors and maternal effect variants in Silver-Russell patients with ICR1 hypomethylation and their mothers. Eur J Hum Genet 27, 42–48 (2019). https://doi.org/10.1038/s41431-018-0269-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-018-0269-1
This article is cited by
-
Trans-acting genetic variants causing multilocus imprinting disturbance (MLID): common mechanisms and consequences
Clinical Epigenetics (2022)
-
The phenotypic variations of multi-locus imprinting disturbances associated with maternal-effect variants of NLRP5 range from overt imprinting disorder to apparently healthy phenotype
Clinical Epigenetics (2019)
