
Abstract
Herein, we describe two members of one family who presented with recurrent episodes of hepatic failure, cerebellar ataxia, peripheral neuropathy, and short stature. Liver transplantation was considered. Whole-exome sequencing (Trio) revealed a synonymous variant in exon 4 of SCYL1:c.459C>T p. (Gly153Gly), which did not appear to affect the protein sequence. Computational prediction analysis suggested that this modification could alter the SCYL1 mRNA splicing processing to create a premature termination codon. The SCYL1 mRNAs in our patient’s lymphocytes were analyzed and aberrant splicing was found. Molecular analysis of family members identified the parents as heterozygous recessive carriers and the proband as well as an affected aunt as homozygous. Evidently, harmless synonymous variants in the SCYL1 gene can damage gene splicing and hence the expression. We confirmed that the pathogenicity of this variant in the SCYL1 gene was associated with spinocerebellar ataxia, autosomal recessive 21 (SCAR21). Other reported cases (accept one) of liver failure found in the SCYL1 variants resolved during childhood, therefore orthotropic liver transplantation was no longer appropriate.
Similar content being viewed by others


Introduction
Spinocerebellar ataxia, autosomal recessive 21 (SCAR21; OMIM #616719) is a very rare subtype of cerebellar ataxia caused by a mutation in the SCYL1 gene. To date, only 11 patients have been described. The phenotype is characterized by recurrent episodes of liver failure resolving during early childhood and hepatic fibrosis. Patients also suffer from neurological symptoms, such as gait disturbances and tremor, as well as peripheral neuropathy and cognitive disability [1, 2].
SCYL1 is a catalytically inactivated protein kinase found in eukaryotic cells and an accessory protein in coatomer I (COPI)-mediated retrograde trafficking from the Golgi apparatus to the endoplasmic reticulum (ER) [3]. A SCYL1 mutation leads to an increase in the volume and surface area of the Golgi apparatus, though its polarity and integrity remain intact. The disruption of Golgi homeostasis is presumed to be caused by a decline in retrograde signaling [4].
The pathophysiology of SCYL1 variants in humans is yet to be resolved. A recent study found that the autosomal recessive mouse neurodegenerative disorder, ‘muscle deficient’ (mdf), is caused by a homozygous 1-bp insertion in exon 8 of the SCYL1 gene, resulting in loss of function. Mdf mice showed progressive neuromuscular atrophy and phenotypes of cerebellar involvement, such as gait ataxia and tremor. Neuropathological examination of the mdf mice noted cerebellar atrophy, Purkinje cell loss, and optic nerve atrophy [5].
Subjects and methods
We describe here two subjects. The first patient was a 28-month-old infant hospitalized in Schneider Children’s Medical Center in Israel and the second patient, his 18-year-old maternal aunt. Whole-exome sequencing was performed after written informed consent was obtained from the proband’s parents and the aunt.
Whole-exome sequencing (centoxome platinium®)
Genomic DNA was isolated from peripheral blood leukocytes according to the standard protocol. Approximately 37 Mb (214,405 exons) of the consensus coding sequences (CCS) were enriched from the fragmented genomic DNA by > 340,000 probes designed against the human genome (Nextera Rapid Capture Exome, Illumina) in addition to the generated library sequenced on an Illumina platform to an average coverage depth of 100–130X. An end-to-end in-house (Centogene) bioinformatic pipeline including base calling, primary filtering of low-quality reads, probable artifacts, and annotation of variants was applied.
All disease-causing variants reported in HGMD®, ClinVar, or CentoMD® (class 1-pathogenic variants), as well as all variants with minor allele frequency (MAF) < 1% in the ExAc database were considered. Evaluation was focused on exons and introns boundaries ± 20. All relevant inheritance patterns were considered. Provided family history and clinical information were used to evaluate the identified variants. Only variants related to the phenotype were reported. Any relevant variant identified by next-generation sequencing (NGS) was in-house validated or Sanger sequenced in the forward and reverse direction to exclude NGS artifacts.
There was no initial assumption as to the mode of inheritance. Given the partial overlapping between the proband’s symptoms and the symptoms associated with pathogenic variants in the SCYL1 gene, mainly liver failure, together with its scarcity in the general population database—minor allele frequency (MAF) of 0.00003, Centogene considered the detected variant as possibly causative.
Splicing evaluation of SCYL1
Total RNA was extracted from the peripheral blood mononuclear cells of the proband, his parents, his aunt, and three controls. Complementary DNA was synthesized, and amplification of SCYL1 was performed by polymerase chain reaction (PCR) using custom-planned primers: forward: 5′-CGTTGGGAATATACCTCAAGGCG-3′, reverse 5′-TGAAGACT TCCCAAATGAGGCAGC-3′. The PCR product was loaded and run through a 1.5% agarose gel. Bands were extracted using a gel clean-up kit (Promega) and sent for confirmation via Sanger sequencing. Sequence alignment was performed using vector NTI software.
This study was approved by the local ethics committee.
Results
Case description
The proband, a 5-month-old infant, was admitted to our hospital due to jaundice and failure to thrive 1 month prior to admission. Jaundice appeared simultaneously with a febrile illness. A significant delay in gross and fine motor development (appropriate for a 2-month-old) was observed. Cognitive milestones were adequate for his age. Perinatal history was unremarkable, except for polyhydramnios and a high head circumference/femur length (HC/FL) ratio, which had appeared on a routine late sonographic follow-up. Parents are non-consanguineous Ashkenazi Jews and the patient is their only child (Fig. 1).

Pedigree in the Jewish Ashkenazi family with SCYL1 variant c.459C>T. Open boxes are asymptomatic family members not tested for carrier status. Dotted boxes are carrier asymptomatic members. The shaded boxes are patients with symptomatic homozygous SCYL1 variants
Family history revealed an 18-year-old maternal aunt with splenomegaly and a history of a transient liver disorder in her infancy (Fig. 1). She has proportional short stature (134.4 cm) and has been followed up through the years due to growth retardation. Major radiographic features reported at age four were delayed bone age, platyspondyly, small femoral epiphyses, flattened and irregular acetabular edges, and cone-shaped epiphyses of the proximal and middle phalanx. These features were not observed in radiographs taken in her adulthood. She also experiences neurological manifestations, such as intention tremor, gait disturbances, progressive stutter, peripheral motor neuropathy, weakness of the upper and lower limbs, and learning disabilities, Table 1. The aunt’s motor functions have been deteriorating and there have been progressive weaknesses of the limbs (first involving proximal muscles). An MRI brain scan performed in her infancy and at age 13 did not demonstrate cerebellar vermis atrophy.
The proband’s physical examination revealed jaundice and hepatosplenomegaly. Laboratory test results showed thrombocytopenia, elevated levels of liver enzymes (AST > ALT), direct hyperbilirubinemia with only slightly elevated GGT, and prolonged INR. Hepatosplenomegaly and early portal hypertension (slow hepatopetal portal flow of 15 cm/s and prominent hepatic artery flow with a resistance index (RI) of 0.86) were observed on sonographic imaging of the abdomen. Liver biopsy, including electron microscopy, was not specific. Pathological changes seen in electron microscopy included distended rough and smooth endoplasmic reticulum, and variable shape deformities of the mitochondria with significant crystolysis and large autophagosomes containing biliary material, especially cytolysis. A thorough laboratory investigation for infectious, metabolic, and immunologic etiologies led to no concrete diagnosis.
During hospitalization, over a course of several days, the proband’s liver functions deteriorated steadily with severe ascites and an episode of bacterial peritonitis. Fortunately, his liver function spontaneously improved and the infant was discharged after 1.5 months, with only mildly elevated liver enzymes, preserved synthetic function, and a slightly decreased platelet count. Over the following months, the infant was admitted twice more due to liver failure. During hospitalization at 1 year of age, a new symmetrical low-frequency action tremor appeared. Neurological examination was otherwise intact. Since the infant presented with recurrent and severe episodes of liver failure, orthotropic liver transplantation was seriously considered. However, before reaching this decision, a whole-exome sequencing (WES) was performed revealing a previously unreported clinically homozygous variant in the SCYL1 gene. This variant was also detected in a homozygous state in the affected aunt.
Molecular investigation
WES (CentoXome PLATINIUM®) was performed on the patient and his parents. A homozygous synonymous substitution in the SCYL1 gene (NM_020680.3;NG_047172.1) c.459C>T;p.(Gly153Gly) chr11:65293678C>T (hg19) located in exon 4 was detectedin the proband. The variant c.459C>T;p. (Gly153Gly) introduces a cryptic splice site within exon 4. Analysis of mRNA confirmed the prediction that this synonymous variant activates a splice-donor site at an early position in exon 4, leading to an out-of-frame mutation with the missing of 145 nucleotides at the 3′ end of exon 4 and protein truncation (p.Glu154Glnfs*66), see supplement. The variant was also detected in both parents of the proband in a heterozygous state. This variant was segregated with the disorder in the family, since it was found in a homozygous state in the affected aunt. Analysis with Alamut software yielded a high prediction rate (5/5 algorithms were positive) that the c.459C>T nucleotide variant could generate a new donor-splice site, thus, disrupting physiological splicing. In addition, the recently published algorithm transcript that inferred the pathogenicity score (TraP) demonstrated a high score for this variant (0.832), implying possible damaging classification [6]. The c.459C>T nucleotide variant has been found only 8 times in over 10,122 alleles of Ashkenazi Jews in the GenomeAD database (allele frequency of ~0.08%) [7].
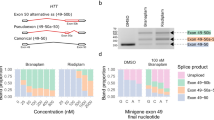
The SCYL1 mRNAs in the patient’s and aunt’s lymphocytes were analyzed and aberrant splicing was found (Fig. 2). Specifically, a point substitution (C > T in exon 4) resulted in an alternative splice site, meaning that the splicing machinery detected the middle of exon 4 as a genuine new splice site and cut the exon in this site. This comes in exchange of further scanning of the exon 4 to reach the wild-type splice site downstream of it. Since the proband and the aunt are homozygous to this variant, only the new splice site was used (hence, one shorter band is visible in Fig. 2). Given that the parents are heterozygous to the variant, the transcript generated both long and short isoforms. This variant was not found in our controls, only the wild-type long transcript was visible. The synonymous variant suggested that no protein structure was affected; however, there were major changes in the mRNA splicing. We note that we did not monitor the protein structures and levels, yet the mRNA sizes indicated a shorter protein formation in the proband's and aunt’s cells (albeit, a mixture of both protein isoforms in the parents).

Variant C > T in exon 4 results in the generation of an alternative splice site. a Simulation of the spliced mRNA structure; boxes and lines represent exons and introns, respectively (not drawn to scale). Exon 4 has an alternative splice site (indicated by an arrow in this and the following panels). b Gel electrophoresis separation of the PCR products from each family member. The splice variants are indicated on the right. c The bottom band from the gel in (b) (marked by an asterisk) was Sanger sequenced and aligned with the UCSC Genome browser. The bottom variant is the mRNA structure taken from the browser, while the upper variant is the aligned actual sequence from the band extracted from the gel. d. A chromatograph of the bands existed from the proband’s DNA sequence
We have submitted the data of the study (variants and phenotypes) into LOVD-Leiden Open Variation Database 3.0. The URL of the database is: https://databases.lovd.nl/shared/ genes/SCYL1 (patient IDs 00164324).
Discussion
We describe herein two members of one family with a homozygous synonymous variant activating a cryptic splice site in the SCYL1 gene. This variant is not as harmless as expected. It is predicted to cause a premature stop codon. Premature translation termination may activate or escape nonsense-mediated decay (NMD), a safeguard mechanism that protects cells from impaired mRNA [8]. We observed the shortened RNA transcript from the mutant allele. It is well known that some premature termination codon (PTC) containing mRNAs can escape NMD [8]. However, the precise molecular mechanism by which the mRNAs escapes, NMD, is poorly understood.
Surprisingly, though the proband and his aunt carry an identical variant, the pedigree revealed no consanguinity, suggesting a founder substitution. Testing for this variant among Ashkenazi Jews with neurodegenerative and hepatic symptomology is warranted.
Skeletal manifestation may represent another phenotype of the SCYL1 variants, as latest reports of patients described multiple skeletal anomalies including short stature, lumbar lordosis, and an abnormal anatomy of bones [2, 9]. Mouse strain SCYL1mdf/SCYL1mdf has also shown dorsoventral flattening of the pelvis by 3 months of age [10]. In our case, the patient’s aunt had short stature, a symptom which was also reported in Lenz et al’s recent publication [2]. We also performed whole-exome sequencing for the aunt to ensure that there was no dual diagnosis, which could explain her skeletal manifestation. No pathogenic variant was found except for the same SCYL1 variant. Interestingly, although carrying the exact same variant as the affected aunt, the proband’s length was normal (7th percentile) and bone age was adequate. However, the proband’s last X-ray, at 25 months, showed an abnormal structure of the thoracic vertebrae. After reviewing the X-rays taken when the proband was 5 months of age, this abnormality was also observed.
Variability in phenotype has been reported in other cases. While one of the families reported by Schmidt et al. was mostly affected by debilitating ataxia due to cerebellar dysfunction, our patient’s aunt was mainly afflicted with motor deterioration due to progressive weakness of the limbs, and families described by Lenz et al. as well as our proband suffered primarily from recurrent and severe episodes of hepatic failure [1, 2]. Variability in phenotype might be partially explained by the location of the mutation in the gene, as early variants have been suggested to cause a wider and more severe phenotype of the syndrome [2].
Furthermore, different tissues are probably more dependent than others on the SCYL1 function. In Pelletier et al’s study, a mouse model with deficient neuronal expression of SCYL1 (NesCre+; Scyl1fl/fl), but preserved skeletal and liver expression presented similar neurodegenerative complaints of the motor neuron system, though the disease was not as severe and progressive as in the Scyl1−/− model (SCYL1-deficient mouse). However, a mouse model with deficient skeletal muscle expression of SCYL1 (CkmmCre+; Scyl1fl/fl) did not develop any disease [11]. The effect of the SCYL1 malfunction on specific phenotypes is not yet understood.
In other humans, findings on a brain MRI performed during late childhood were suggestive of cerebellar vermis atrophy [1]. Notwithstanding a new complaint of tremor suspected of cerebellar origin appeared in our patient, brain MRI did not reveal any cerebellar abnormalities. A lag in time between the atrophic findings on imaging and clinical complaints might explain this discrepancy. A future follow-up as to the development of pathological findings on brain imaging will definitely help in clarifying this matter.
The infant was 28 months at his last examination. The last episode of liver dysfunction, fifth in number, occurred at 25 months. Interestingly, deterioration in liver function did not occur following every single febrile illness, and there was no other apparent pattern of the trigger. Currently, the patient’s major complaints are tremor mainly on action and gait disturbances. Developmental milestones were delayed and were appropriate for an 18–20 month-old child. Growth parameters improved following percutaneous endoscopic gastrostomy (PEG) insertion. Only one of the previously reported patients underwent liver transplantation [2]. Our patient’s liver functions stabilized, hence liver transplantation was not performed.
This is the first case description of a synonymous variant in SCYL1 pathogenetics. Synonymous nucleotide substitutions in gene coding regions which do not alter protein sequences were historically thought to have no effect and were thus called ‘silent variants’. This belief has been overturned in recent years, as synonymous variants are now widely acknowledged to cause diseases [12, 13]. It is clear that synonymous variants should not be filtered out if no other clear-cut mutations are found in the causative genes. Bioinformatics software such as the TraP score (http://trap-score.org) for predicting splice sites may be an efficient tool for identifying potentially damaging synonymous variants [6]. We believe that an assay based on the patient’s blood cells and controls could be designed to confirm aberrant splicing caused by synonymous variants.
Conclusion
Variants in the SCYL1 gene might be involved in multi-organ dysfunction, mainly affecting the liver and nervous systems. There is significant variability in phenotypes, even between members of the same family carrying an identical variant. Furthermore, clinical signs vary with age. Mutations in the SCYL1 gene should be included in the differential diagnosis of patients with impaired liver function and neurological symptoms, particularly if cerebellar involvement is suspected. This case emphasizes the importance of synonymous variants in the pathogenesis of the disease. Clinical correlation and functional analysis are required to confirm the diagnosis.
Web Resources
The URL for data presented here are as follows: Exac Browser Data, http://exac.broadinstitute.org/; Transcript-inferred pathogenicity score, http://trap-score.org/about.jsp
References
Schmidt WM, Rutledge SL, Schüle R, Mayerhofer B, Züchner S, Boltshauser E, et al. Disruptive SCYL1 mutations underlie a syndrome characterized by recurrent episodes of liver failure, peripheral neuropathy, cerebellar atrophy, and ataxia. Am J Hum Genet. 2015;97:855–61.
Lenz D, McClean P, Kansu A, Bonnen PE, Ranucci G, Thiel C, et al. Scyl1 variants cause a syndrome with low γ-glutamyl-transferase cholestasis, acute liver failure, and neurodegeneration (CALFAN). Genet Med 2018; epub ahead of print8 February 2016; https://doi.org/10.1038/gim.2017.260.
Burman JL, Bourbonniere L, Philie J, Stroh T, Dejgaard SY, Presley JF, et al. Scyl1, mutated in a recessive form of spinocerebellar neurodegeneration, regulates COPI-mediated retrograde traffic. J Biol Chem. 2008;283:22774–86.
Burman JL, Hamlin JN, McPherson PS. Scyl1 regulates Golgi morphology. PLoS ONE. 2010;5:e9537. (2010)
Schmidt WM, Kraus C, Höger H, Hochmeister S, Oberndorfer F, Branka M, et al. Mutation in the Scyl1 gene encoding amino-terminal kinase-like protein causes a recessive form of spinocerebellar neurodegeneration. EMBO Rep. 2007;8:691–7.
Gelfman S, Wang Q, McSweeney KM, Renz Z, La Carpia F, Halvorsen M, et al. Annotating pathogenic non-coding variants in genic regions. Nat Commun. 2017;8:236.
Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–91.
Kurosaki T, Maquat LE. Nonsense-mediated mRNA decay in humans at a glance. J Cell Sci. 2016;129:461–7.
Smith ED, Radtke K, Rossi M, et al. Classification of genes: standardized clinical validity assessment of gene-disease associations aids diagnostic exome analysis and reclassifications. Hum Mutat. 2017;38:600–8.
Blot S, Poirier C, Dreyfus PA. The mouse mutation muscle deficient (mdf) is characterized by a progressive motoneuron disease. J Neuropathol Exp Neurol. 1995;54:812–25.
Pelletier S, Gingras S, Howell S, Vogel P, Ihle JN. An early onset progressive motor neuron disorder in Scyl1-deficient mice is associated with mislocalization of TDP-43. J Neurosci. 2012;32:16560–73.
Hunt RC, Simhadri VL, Iandoli M, Sauna ZE, Kimchi-Sarfaty C. Exposing synonymous mutations. Trends Genet. 2014;30:308–21.
Sauna ZE, Kimchi-Sarfaty C. Understanding the contribution of synonymous mutations to human disease. Nat Rev Genet. 2001;12:683–91.
Acknowledgements
We thank all the family members described in this paper for their participation in this study. The authors thank Mrs. Phyllis Curchack Kornspan for her editorial services.
Funding
The Shomron team was partially funded by the Adelis Foundation and the Israel Science Foundation (ISF, 1852/16).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Shohet, A., Cohen, L., Haguel, D. et al. Variant in SCYL1 gene causes aberrant splicing in a family with cerebellar ataxia, recurrent episodes of liver failure, and growth retardation. Eur J Hum Genet 27, 263–268 (2019). https://doi.org/10.1038/s41431-018-0268-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-018-0268-2
This article is cited by
-
Whole exome sequencing in Alopecia Areata identifies rare variants in KRT82
Nature Communications (2022)
-
mTORC1 controls Golgi architecture and vesicle secretion by phosphorylation of SCYL1
Nature Communications (2022)
-
Lipid Dyshomeostasis and Inherited Cerebellar Ataxia
Molecular Neurobiology (2022)
-
The intronic branch point sequence is under strong evolutionary constraint in the bovine and human genome
Communications Biology (2021)
-
The Classification of Autosomal Recessive Cerebellar Ataxias: a Consensus Statement from the Society for Research on the Cerebellum and Ataxias Task Force
The Cerebellum (2019)
