
Abstract
In oncology, the expanding use of multi-gene panels to explore familial cancer predisposition and tumor genome analysis has led to increased secondary findings discoveries (SFs) and has given rise to important medical, ethical, and legal issues. The American College of Medical Genetics and Genomics published a policy statement for managing SFs for a list of genes, including 25 cancer-related genes. Currently, there are few recommendations in Europe. From June 2016 to May 2017, the French Society of Predictive and Personalized Medicine (SFMPP) established a working group of 47 experts to elaborate guidelines for managing information given on the SFs for genes related to cancers. A subgroup of ethicists, lawyers, patients’ representatives, and psychologists provided ethical reflection, information guidelines, and materials (written consent form and video). A subgroup with medical expertise, including oncologists and clinical and molecular geneticists, provided independent evaluation and classification of 60 genes. The main criteria were the “actionability” of the genes (available screening or prevention strategies), the risk evaluation (severity, penetrance, and age of disease onset), and the level of evidence from published data. Genes were divided into three classes: for class 1 genes (n = 36), delivering the information on SFs was recommended; for class 2 genes (n = 5), delivering the information remained questionable because of insufficient data from the literature and/or level of evidence; and for class 3 genes (n = 19), delivering the information on SFs was not recommended. These guidelines for managing SFs for cancer-predisposing genes provide new insights for clinicians and laboratories to standardize clinical practices.
Similar content being viewed by others

Introduction
The expansion in panel, exome, and genome analyses by next-generation sequencing (NGS) for clinical purposes has led to an increase in secondary findings (SFs). SFs are results of a deliberate or incidental screening for alterations in genes that are not relevant to the diagnostic indication for which the sequencing test was ordered [1, 2], unlike primary findings that are used to describe pathogenic alterations in genes that are relevant to the diagnosis.
In oncology, the use of multi-gene panel analyses to explore familial cancer syndrome has expanded greatly and has led to the discovery of gene alterations that are not directly related to the initial spectrum of the search [3,4,5,6,7,8]. Somatic tumor panels or genome analysis can also lead to germline SFs for cancer genes. In exome analysis, the rate of actionable genetics SFs was estimated from 2 to 5% [9,10,11]. This increased detection of potentially clinically significant heritable deleterious variants over the predicted yield of targeted germline testing has given rise to important medical, ethical and legal issues on prevention for patients and their families.
The American College of Medical Genetics and Genomics (ACMG) published a clear policy statement related to appropriate information about the possibility of secondary results given in consultation before testing and recommended that laboratories report pathogenic alterations for a list of genes, of which 25 (23 in the previous version) were cancer-predisposing genes [2, 12, 13]. Currently, there are few equivalent recommendations in Europe to manage information given on SFs for cancer-related genes (those of the European Society of Human Genetics [ESHG] [14] and those of the “100,000 Genomes Project”: https://www.genomicsengland.co.uk/taking-part/results/). Recent publications have shown an important variability in European practices for the transmission of SF information from the laboratory to the clinician [15] and from the clinician to the patient [16]. Thus, we urgently need guidelines to help European molecular laboratories and clinician physicians harmonize their practice, particularly for cancer-related genes [14, 17].
The French Society of Predictive and Personalized Medicine (Société Française de Médecine Prédictive Personnalisée [SFMPP]) set up a multidisciplinary in-depth reflection on the medical, ethical, and legal issues related to SFs for cancer-associated genes in order to elaborate guidelines for managing information given on SFs for cancer-related genes. Here, we describe the process of this reflection and mainly describe the classification system resulting from the evaluation of genes by the medical subgroup in terms of information given on SFs. We also present the guidelines and materials (informed consent forms and information media tools) for the clinical management of SFs for cancer-related genes.
Materials and methods
Composition of the expert panels
The SFMPP is an independent learned society funded by donations from public support. The SFMPP gathered a group of 47 multidisciplinary experts divided into two working subgroups. The first subgroup addressed ethical and legal questions and the second medical expertise. The ethical and legal group (n = 17) consisted of three psychologists, one sociologist, four ethicists, two methodologists, four lawyers, and three representatives of patient associations (BRCA France, the Vaincre les Maladies Lysosomales association and the Association pour la Prévention, Traitement Etude des Polyposes Familiales). The objective of the ethical and legal subgroup was to elaborate general recommendations on information related to patients and consent and to provide informed consent forms and an information media tool (animation movie, presented in Supplementary Information). The medical expertise group (n = 30) consisted of eight oncologists, 11 clinical geneticists, three molecular biologists, and eight cancer gene experts. The objective for the medical subgroup was to provide independent evaluation and classification of cancer genes for cancer risk and actionability according to the criteria defined in the Methods section.
Genes evaluated
The medical subgroup defined a primary list of 80 cancer-related genes for which inherited deleterious variants have been shown to predispose to cancer. This list of candidate genes (Table 1) was established with a search of the literature and the professional experience of the steering committee. The gene NF2 was excluded from the list before evaluation because of its association with a non-malignant tumor phenotype, unlike NF1, which predisposes to malignant peripheral nerve-sheath tumors. Also, 19 genes responsible for pediatric-onset cancer predisposition (DICER1, SMARCB1, SMARCA4, RB1, ALK, WT1, PRKAR1A, PTCH1, PTCH2, XP genes PTCH1 and PTCH2, XP genes [XPA to XPG including XPV], SUFU, PRSS1, SUFU, PRSS1) were excluded (but not TSC1 and TSC2 for which predisposing to renal cell carcinoma most often occurs in the adult setting [18]) because the present work aimed to elaborate recommendations for adults. This choice was justified by the lack of involvement of the child in the genetic testing process (subject to parental agreement), the sensitive character that can take such announcements and the fact that most frequent cancer predispositions affect adults. Thus, this topic for children will require specific considerations.
The actionability assessment
A total of 60 genes were evaluated and scored by the SFMPP medical subgroup. The genes were classified by risk and actionability (validated screening or prevention strategies available) into three classes according to the criteria described below. Finally, 228 evaluation sheets were completed by 16 independent experts and analyzed by the steering committee. The evaluation methodology and a flowchart of the evaluation process is in Supplementary Information. Briefly, the group evaluated genes in five meetings from June 2016 to May 2017 (held in Paris, Montpellier, and Dijon, France). After a synthesis of the existing literature at the time of the evaluation, each gene was evaluated for risk (severity, penetrance, age of disease onset) and actionability as well as level of evidence of this information in the literature. A mean of 2.65 experts evaluated each gene. Discrepancies in evaluation were discussed with the steering committee.
The evaluation questionnaire is presented in Table 2 and the evaluation process is presented in Fig. 1. To strengthen the evaluation score for the perspective of actionability, the penetrance score was adjusted (doubled) for power. Thus, early predispositions, justifying the earliest actionability, were highlighted. If the gene concerned predisposition to several tumor pathologies, the evaluators were asked to assess each risk when possible. The mean value of the evaluation score was retained for each gene. Of note, these recommendations apply to anomalies of a certain deleterious nature (pathogenic variant [19], class 5 [20] of the International Agency for Cancer Research [IARC]) affecting the function of the protein, consistent with Human Genome Variation Society nomenclature.

SFMPP working method. The working method of the Society of Predictive and Personalized Medicine (SFMPP) was adapted from the American College of Medical Genetics and Genomics methodology, to classify the “actionable” cancer-related genes for adults. ClinGen AWG Clinical Genome Resource Actionability Working Group (USA), WGGP Working Group on Gene Phenotype
Classification
The medical group classified the remaining 60 genes into three classes shown in Table 3. Class 1 includes “actionable” genes for which informing the clinician and the patient is recommended because of available screening or preventive strategies. Class 2 includes genes with significant risk and screening or preventive measures, but the literature data or level of evidence at this time are not sufficient to consider the real benefit of an intervention. Class 3 includes genes with moderate risk of cancer and/or with limited or non-existence screening and prevention strategies, for which informing patients is not recommended. Some genes were classified in class 3 because of an insufficient actionability score (e.g., FLNC, MC1R, MITF) or low level of evidence or data in the literature (e.g., GREM1, NTHL1, MDH2, SDHAF2) [21,22,23].
Results
Classification of cancer genes according to their actionability
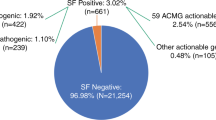
The list of actionable genes is presented in Table 1 (their SFMPP score is available in Supplementary Information) and compared to the ACMG classification (updated) in Fig. 2.

Comparison of the SFMPP and ACMG scoring of actionable cancer-genes. For the SFMPP classification, classes 1, 2, and 3 are in red, black, and blue, respectively. Actionable genes for SFMPP (blue circle) and the ACMG classification (SF v1.0, green circle, and SF v2.0, gray circle) are represented. hmz homozygote
Synthesis of recommendations
The SFMPP recommendations on SFs in cancer-related genes are as follows:
-
(1)
Patients should be informed of SFs for actionable predisposition genes related to hereditary cancers (class 1 genes, Table 1) if written informed consent concerning SFs was previously given.
-
(2)
Independent of the primary indication of the genetic analysis, if SFs are identified in actionable cancer-predisposition genes (class 1 genes), they should be reported by the laboratory to the clinician.
-
(3)
Fair and clear information on SFs in actionable cancer-predisposition genes should be provided to the patient before every sample collection for a panel, exome or genome sequencing, for both constitutive or somatic analysis.
-
(4)
Information must be provided and informed consent collected by a health professional qualified in medical genetics (oncogeneticist, geneticist, oncologist and gynecologist trained in oncogenetics, genetic counselors).
-
(5)
The patient’s autonomy and desire to know or to ignore such SF results must be respected. Patients could decline at any time to be informed about these SFs, even if they previously gave their approval.
-
(6)
Suitable standardized and specific information support for patients must be created and shared.
-
(7)
A specific informed consent form specifying the desire to be or not be informed about relevant SFs should be collected:
-
At the initial prescription for the analysis
-
At the announcement of the primary analysis result, to confirm the patient’s desire for SF information independent of the primary analysis results (confirmatory consent).
-
(8)
Information and informed consent must mention the medical impact on relatives that SFs can represent. Standardized information tools such as digital or educational support may be encouraged.
-
(9)
SF results must be announced by a doctor qualified in medical genetics who is a member of a multidisciplinary team qualified for pre-symptomatic diagnosis.
-
(10)
A systematic collection of informed consent about constitutional SFs is recommended when prescribing a genetic analysis for tumor (somatic) characteristics for therapeutic purposes.
The gene list should be regularly updated according to the evolution of knowledge about the cancer risk, the actionability and the level of evidence.
Patient information and autonomy
The SFMPP supports the need to improve and standardize the medical information related to SFs for the patient. Therefore, we developed a digital support with a collaborative partnership between the FHU-TRANSLAD federation, AnDDI-rare pathway, the Centre de reference anomalies du développement and the SFMPP for supporting giving information to patients as a 5-minute video entitled “Secondary data from high throughput genome sequencing” (in French with English subtitles). To improve the quality and harmonization of information given to the patient, this media tool could provide important and consistent support to communicate with the patient.
The patient’s autonomy and desire to know or ignore such results is a major point that must be respected. Based on the principle of benefit, autonomy and respect of the right not to know, the SFMPP ethical and legal subgroup elaborated specific consent forms (in Supplementary Information) for genomic studies. These consent forms can be adapted for clinical or research genomic analysis. The working group recommends collecting a first informed consent about SFs during the initial medical procedure motivating the primary genetic analysis. A second informed consent form (Fig. 3) is offered after the announcement of the primary findings so that the patient could, with more autonomy, differentiate the issues and confirm or refuse access to this information. The delivery of the SF results should be the subject of a dedicated consultation. The need to dissociate the announcement of the primary findings from that of SFs was supported by patient associations to limit the psychological impact generated.

Recommended steps for reporting secondary findings to the patient
The information concerning a multiple-gene analysis using NGS differs from that provided for targeted genetics testing because it must consider the possibility of secondary data discovery. Although detailing all eventualities of incidental discoveries when informing the patient is impractical, the patient should be provided with the main elements to allow for an informed choice when giving consent.
The clinical utility (in terms of screening and prevention), for personal and family reasons, of a secondary discovery of a pathogenic variant within an actionable gene must be explained to the patient and counterbalanced by the arguments against this information. Informing patients’ relatives about genetic discovery of a primary or secondary variant in actionable genes is not only a moral responsibility but, in terms of French law, also represents a legal duty for the proband [24]. The disclosure to relatives can be particularly painful for families, even deleterious in some cases, but how this information is given to relatives should be included in the informed consent to allow the patient a better understanding and consideration of the implications of results. To facilitate this dissemination, the medical genetics team can disclose the result to the family if the patient so authorizes, thereby protecting as much as possible the patient’s anonymity.
Limitations and interpretation of SFs
We deliberately chose to restrict these recommendations to deleterious variants of cancer-related genes (pathogenic variant [19], IARC class 5 [20]). Thus, IARC class 4 variants (probably pathogenic) and class 3 variants (variants of uncertain significance) are not retained for these guidelines. Particularly for SFs, limiting actionability to clearly pathogenic variants seems reasonable. Information on IARC variants of classes 1 (benign) and 2 (likely benign) should not be given to the practitioner or the patient because these results, unnecessary for the patient’s healthcare, can involve misinterpretation of the results.
The SFMPP admits that the sensitivity of variant detection may vary among genes according to the coverage and depth of the sequencing process. Thus, the coverage and depth at the position of the gene of interest will not be the same as that of genes secondarily and deliberately studied. This concern could result in misinterpretation by the patient and/or clinician of the results, such as the absence of a pathogenic variant in secondarily studied genes. Therefore, the report should mention the conditions of sequencing and analysis of secondary data if it differs from that of the primary data. Hence, clinicians may inform the patient that not all inherited predispositions to cancer can be excluded if no SF is detected.
The lack of detection of pathogenic variants in genes secondarily explored particularly applies to genomic analyses carried out for research purposes. The SFMPP recognizes that genomic analysis performed for health research does not always have the same sensitivity and quality criteria (depth and coverage) as that performed in a healthcare context. The lack of detection of a pathogenic variant in a secondary gene cannot be the responsibility of the laboratory (in a healthcare context or research protocol) because the technique does not have the necessary power to highlight it.
For SFs, the mention of the arguments (literature and in silico scoring) allowing to establish the pathogenicity of the reported secondary variant should be mentioned in the analysis report sent to the clinician to harmonize the understanding of the results. Given the possibility for patients to reconsider their decision to access SF results at the time of the results of the primary analysis, SF results should be reported in a second report independent of the primary findings. The re-analysis of previously generated sequencing data leading to the discovery of SFs is also completely relevant to these guidelines. The SFMPP does not advocate or oppose the active search for SFs in genes included in the SFMPP list when performing genomic examination and considers that this dynamic depends on the policy of each laboratory or medical structure with clear informed consent of the patient.
Somatic sequencing in tumors or other human tissues
Somatic multi-gene analysis with NGS can reveal germline deleterious variants as SFs [25, 26]. The need for significant sequencing depth for these somatic analyses implies that the germline characteristic of a pathogenic variant (IARC class 5) will be suspected even before confirmation by constitutional analysis. The absence of prior information regarding the search for constitutional SFs and the lack of patient positioning by informed consent are problematic. The SFMPP recommends that such analyses be the subject of a written informed consent form appropriate to the discovery of SFs before somatic analysis and before the return of results. These recommendations concern both somatic analyses performed in a healthcare context or for research. Particularly, for research purposes, the consent form should be written or validated by a medical ethics committee before being offered to the patient.
Discussion
The expanding use of multiple panel analysis at constitutional or somatic levels in clinical practice leads to frequent discoveries of SFs for cancer-related genes. The ACMG published a clear policy statement related to appropriate information about the possibility of secondary results given in consultation before testing, but recommendations were lacking in Europe. Here, the French SFMPP presents recommendations for managing SFs for cancer-related genes in adults.
The work provided by the SFMPP is comprehensive and relevant for the patient, the laboratory and the clinician. Therefore, we needed to ensure the multidisciplinary composition of the working groups. The guidelines of the SFMPP are expressed in a context of a European extensive genome project. Indeed, several European states such as the UK (100,000 Genomes Project) [27, 28], Iceland [29,30,31], Estonia (Estonian Genome Project) [32,33,34], France (Genomic Medicine France 2025) [35], The Netherlands (Genome of The Netherlands Project) [36], and Germany (National Genome Research Network) [37] have drawn up government plans to develop and generalize the use of large-scale genetic analyses. In this dynamic, it is likely that SFs will be more frequent and a harmonization of practices to better care for the patient seems necessary more than ever.
The SFMPP admits that the use of an evaluation questionnaire, even if evaluated by several recognized experts, may represent an evaluator-dependent subjectivity bias even with a mean of 2.65 experts evaluating each gene. To limit this bias and to globally homogenize the results, evaluation criteria were designed on the models of the ACMG and the Clinical Genome Resource (ClinGen) [12, 38, 39]. We aimed to develop an open, reusable questionnaire for future SFMPP topics including genes not related to cancer predisposition. Therefore, the questionnaire does not detail the tumor pathology concerned.
The “actionability” term should be discussed, because intentionally we did not consider it from the viewpoint of isolated genetic counseling (to relatives or for prenatal purposes). For example, the discovery of a heterozygous MUTYH variant affecting protein function would essentially lead to genetic counseling but would have little impact on the patient’s care. Hence, consent for SFs would not then fully correspond to the result. In the same way, the psychological impact of such results on the patient is not negligible with limited actionability for the patient.
The SFMPP recommendations may apply to genomic research. Research studies of constitutional genomics or somatic genetics should be adapted to the written informed consent as for genomic analyses performed in a medical context.
Although, there is an important overlap between the ACMG and SFMPP lists of actionable genes, additional genes are classified as class 1, requiring a return to the patient according to the SFMPP. This situation might be due to increased knowledge on risk estimation and/or validation of screening or prevention strategies. For instance, PALB2 has been recently confirmed as a major predisposing gene for breast cancer.
Recent studies have shown that RNA-seq has an efficiency comparable to that of whole-exome sequencing on tumor tissue in the detection of somatic genetic variants [40]. Although these transcriptomic analyses of tumor tissue are outside the scope of these recommendations, they could lead secondarily to the identification of germline variants in cancer-susceptibility genes and constitute SFs. Thus, before performing these analyses, the patient could be proposed a written informed consent for SFs, like that for somatic genetic analyses.
Personalized therapies are conditioned by the presence of a mutated gene of interest in the tumor but also at the constitutional level. This is the case for poly(ADP) ribose polymerase (PARP) inhibitors such as olaparib for metastatic ovarian cancer in patients with mutations in BRCA1 and BRCA2 [41, 42]. This was also the case in the study of the anti-programmed cell death 1 antibodies developed for hypermutated tumors linked, among others, to mismatch repair deficiency [38, 43,44,45]. Cancer-related genes account for a large part of the actionable genes.
The steering committee decided to limit these recommendations to SFs for cancer-related genes in adults. Indeed, most hereditary tumor syndromes occur only in adulthood. Because of the lack of involvement of children in the process of genetic analysis (decision by written consent of parents or legal guardians) and the lack of maturity of children in the SF perspective, these recommendations for children are both sensitive and complex. In terms of actionable tumor-predisposition genes in children, a dedicated subgroup should be created separate from the development of the current guidelines.
One of the most important challenges for the ethical and legal subgroup was to define guidelines to manage patient information and the use of written consent by focusing on three major principles: the medical benefit, the autonomy of the patient and the right to know and not to know. Our dedicated working group on information and consent started with a review of available informed consents in Europe. Some of our members from Switzerland and France already participated in the elaboration of national informed consent in genetics (French Genetic and Cancer group, INSERM Ethics group). For SFs in cancer genes, we have built a specific consent taking into account available works, legal aspects, and patient association considerations. The use of double consent, allowing for a period of reflection and the opportunity for patients to express their opinion again, seemed an important element, not found in the literature to our knowledge. The second SF consent, proposed during the reporting of primary findings, should also be offered again to patients who have stated in the first consent that they did not want to know the SFs. Thus, as for all patients, these patients will have the right to return to the decision after a period of reflection. Even if no formal pilot experiment of such a double consent was performed by the group, this is in accordance with the dynamic consent approach recently proposed to provide adaptive consent for research [46]. To improve the quality and harmonization of information given to the patient, we believe that the video tool could provide important and consistent support for communicating with the patient.
The present recommendations are not meant to substitute for the clinical and ethical judgement of clinician, but they are proposed as a basis for reflection to help with the prescription and the results of the genomic analyses in a homogeneous way. These recommendations are not a substitute for a personal bibliographic watch that is an integral part of the clinician’s work in a context of continuous improvement of medical knowledge due to the variety of cancer-predisposition syndromes and the technical complexity. Thus, these guidelines cannot be held responsible for the management of the SFs, which is the responsibility of the clinician.
The list of genes established by the SFMPP is not exclusive; it is considered a “minimal list” of actionable genes predisposing to monogenic cancer in adults. Furthermore, for SF management, likely pathogenic (IARC class 4) [19, 20] variants and variants of uncertain significance (IARC class 3) seemed too uncertain for the SFMPP workgroup for consensus. The implementation of large-scale sequencing in current practice (and in the general population) will probably redefine the frequency and pathogenicity of such variants.
Because of the evolution of knowledge, the reclassification of variants of uncertain significance or likely pathogenic variants to pathogenic variants could lead to a return of results representing SFs. Therefore, the management of such SFs by reclassification could be oriented by the position of the patient about these results in the two informed consent forms previously signed.
Similarly, the current recommendations are developed according to the state of knowledge at the time of writing, but they are expected to evolve. A periodic review of SFMPP genes should allow for reclassifying genes with higher proof.
Regarding the future perspectives of the SFMPP, it seems necessary, in parallel with this literature watch, to establish a monitoring of SF census at the European level to better adapt the current recommendations to laboratory and clinicians’ practices [47].
Managing SFs associated with cancer-related genes has become an emerging concern for clinicians and laboratories because of the extensive use of gene panels and large-scale genomic analysis at somatic and germline levels. This work provides a first step toward standardized guidelines in France and Europe for SFs related to cancer-predisposing genes.
References
Weiner C. Anticipate and communicate: ethical management of incidental and secondary findings in the clinical, research, and direct-to-consumer contexts (December 2013 report of the Presidential Commission for the Study of Bioethical Issues). Am J Epidemiol. 2014;180:562–4.
Kalia SS, Adelman K, Bale SJ, Chung WK, Eng C, Evans JP, et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SFv2.0): a policy statement of the American College of Medical Genetics and Genomics. Genet Med. 2017;19:249–55.
Tung N, Lin NU, Kidd J, Allen BA, Singh N, Wenstrup RJ, et al. Frequency of germline mutations in 25 cancer susceptibility genes in a sequential series of patients with breast cancer. J Clin Oncol. 2016;34:1460–8.
O’Leary E, Iacoboni D, Holle J, Michalski ST, Esplin ED, Yang S, et al. Expanded gene panel use for women with breast cancer: identification and intervention beyond breast cancer risk. Ann Surg Oncol. 2017;24:3060–6.
Stafford JL, Dyson G, Levin NK, Chaudhry S, Rosati R, Kalpage H, et al. Reanalysis of BRCA1/2 negative high risk ovarian cancer patients reveals novel germline risk loci and insights into missing heritability. PLoS One. 2017;12:e0178450.
Aloraifi F, McDevitt T, Martiniano R, McGreevy J, McLaughlin R, Egan CM, et al. Detection of novel germline mutations for breast cancer in non-BRCA1/2 families. FEBS J. 2015;282:3424–37.
Gieldon L, Masjkur JR, Richter S, Darr R, Lahera M, Aust DE, et al. Next generation panel sequencing identifies NF1 germline mutations in three patients with pheochromocytoma but no clinical diagnosis of Neurofibromatosis Type 1. Eur J Endocrinol. 2017;178:K1–9.
Ring KL, Bruegl AS, Allen BA, Elkin EP, Singh N, Hartman AR, et al. Germline multi-gene hereditary cancer panel testing in an unselected endometrial cancer cohort. Mod Pathol. 2016;29:1381–9.
Amendola LM, Dorschner MO, Robertson PD, Salama JS, Hart R, Shirts BH, et al. Actionable exomic incidental findings in 6503 participants: challenges of variant classification. Genome Res. 2015;25:305–15.
Dorschner MO, Amendola LM, Turner EH, Robertson PD, Shirts BH, Gallego CJ, et al. Actionable, pathogenic incidental findings in 1,000 participants’ exomes. Am J Hum Genet. 2013;93:631–40.
Dewey FE, Murray MF, Overton JD, Habegger L, Leader JB, Fetterolf SN, et al. Distribution and clinical impact of functional variants in 50,726 whole-exome sequences from the DiscovEHR study. Science. 2016;354:aaf6814.
Green RC, Berg JS, Grody WW, Kalia SS, Korf BR, Martin CL, et al. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet Med. 2013;15:565–74.
Directors ABo. ACMG policy statement: updated recommendations regarding analysis and reporting of secondary findings in clinical genome-scale sequencing. Genet Med. 2015;17:68–9.
van El CG, Cornel MC, Borry P, Hastings RJ, Fellmann F, Hodgson SV, et al. Whole-genome sequencing in health care: recommendations of the European Society of Human Genetics. Eur J Hum Genet. 2013;21:580–4.
Vears DF, Senecal K, Borry P. Reporting practices for unsolicited and secondary findings from next-generation sequencing technologies: perspectives of laboratory personnel. Hum Mutat. 2017;38:905–11.
Hehir-Kwa JY, Claustres M, Hastings RJ, van Ravenswaaij-Arts C, Christenhusz G, Genuardi M, et al. Towards a European consensus for reporting incidental findings during clinical NGS testing. Eur J Hum Genet. 2015;23:1601–6.
Claustres M, Kozich V, Dequeker E, Fowler B, Hehir-Kwa JY, Miller K, et al. Recommendations for reporting results of diagnostic genetic testing (biochemical, cytogenetic and molecular genetic). Eur J Hum Genet. 2014;22:160–70.
Yang P, Cornejo KM, Sadow PM, Cheng L, Wang M, Xiao Y, et al. Renal cell carcinoma in tuberous sclerosis complex. Am J Surg Pathol. 2014;38:895–909.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24.
Plon SE, Eccles DM, Easton D, Foulkes WD, Genuardi M, Greenblatt MS, et al. Sequence variant classification and reporting: recommendations for improving the interpretation of cancer susceptibility genetic test results. Hum Mutat. 2008;29:1282–91.
Cascon A, Comino-Mendez I, Curras-Freixes M, de Cubas AA, Contreras L, Richter S, et al. Whole-exome sequencing identifies MDH2 as a new familial paraganglioma gene. J Natl Cancer Inst. 2015;107.
Bausch B, Schiavi F, Ni Y, Welander J, Patocs A, Ngeow J, et al. Clinical characterization of the pheochromocytoma and paraganglioma susceptibility genes SDHA, TMEM127, MAX, and SDHAF2 for gene-informed prevention. JAMA Oncol. 2017;3:1204–12.
Bayley JP, Kunst HP, Cascon A, Sampietro ML, Gaal J, Korpershoek E, et al. SDHAF2 mutations in familial and sporadic paraganglioma and phaeochromocytoma. Lancet Oncol. 2010;11:366–72.
Touraine M. Décret no. 2013-527 relatif aux conditions de mise en œuvre de l’information de la parentèle dans le cadre d’un examen des caractéristiques génétiques à finalité médicale; in: santéMdasedl (ed), 20 juin 2013.
Abdel-Rahman MH, Rai K, Pilarski R, Davidorf FH, Cebulla CM. Germline BAP1 mutations misreported as somatic based on tumor-only testing. Fam Cancer. 2016;15:327–30.
Bijlsma RM, Bredenoord AL, Gadellaa-Hooijdonk CG, Lolkema MP, Sleijfer S, Voest EE, et al. Unsolicited findings of next-generation sequencing for tumor analysis within a Dutch consortium: clinical daily practice reconsidered. Eur J Hum Genet. 2016;24:1496–500.
Peplow M. The 100,000 Genomes Project. BMJ. 2016;353:i1757.
Samuel GN, Farsides B. Public trust and ‘ethics review’ as a commodity: the case of Genomics England Limited and the UK’s 100,000 genomes project. Med Health Care Philos. 2018;21:159–68.
Greely HT. Iceland’s plan for genomics research: facts and implications. Jurimetrics. 2000;40:153–91.
Gudbjartsson DF, Helgason H, Gudjonsson SA, Zink F, Oddson A, Gylfason A, et al. Large-scale whole-genome sequencing of the Icelandic population. Nat Genet. 2015;47:435–44.
Palsson G, Rabinow P. Iceland: the case of a national human genome project. Anthropol Today. 1999;15:14–8.
Sutrop M, Simm K. The Estonian healthcare system and the genetic database project: from limited resources to big hopes. Camb Q Healthc Ethics. 2004;13:254–62.
Metspalu A, Estonian Genome Project—before the take-off and take-off. Bioinformatics. 2002;18(Suppl 2):S152
Metspalu A,Kohler F,Laschinski G,Ganten D,Roots I, [The Estonian Genome Project in the context of European genome research]. Dtsch Med Wochenschr. 2004;129(Suppl 1):S25–8.
Levy Y. Genomic medicine 2025: France in the race for precision medicine. Lancet. 2016;388:2872.
Boomsma DI, Wijmenga C, Slagboom EP, Swertz MA, Karssen LC, Abdellaoui A, et al. The Genome of the Netherlands: design, and project goals. Eur J Hum Genet. 2014;22:221–7.
Bickeboller H. [The National Genome Research Network. Genome research in Germany]. Bundesgesundheitsblatt Gesundh Gesundh. 2007;50:168–73.
Rehm HL, Berg JS, Brooks LD, Bustamante CD, Evans JP, Landrum MJ, et al. ClinGen—the clinical genome resource. N Engl J Med. 2015;372:2235–42.
Strande NT, Riggs ER, Buchanan AH, Ceyhan-Birsoy O, DiStefano M, Dwight SS, et al. Evaluating the clinical validity of gene-disease associations: an evidence-based framework developed by the clinical genome resource. Am J Hum Genet. 2017;100:895–906.
Van Allen EM, Robinson D, Morrissey C, Pritchard C, Imamovic A, Carter S, et al. A comparative assessment of clinical whole exome and transcriptome profiling across sequencing centers: implications for precision cancer medicine. Oncotarget. 2016;7:52888–99.
Nirsimloo R, Gourley C. The safety and efficacy of olaparib therapy in patients with relapsed ovarian cancer. Expert Rev Anticancer Ther. 2016;16:597–603.
Tewari KS, Eskander RN, Monk BJ. Development of olaparib for BRCA-deficient recurrent epithelial ovarian cancer. Clin Cancer Res. 2015;21:3829–35.
Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science. 2017;357:409–13.
Gong J, Cho M, Sy M, Salgia R, Fakih M. Molecular profiling of metastatic colorectal tumors using next-generation sequencing: a single-institution experience. Oncotarget. 2017;8:42198–213.
Gong J, Wang C, Lee PP, Chu P, Fakih M. Response to PD-1 blockade in microsatellite stable metastatic colorectal cancer harboring a POLE mutation. J Natl Compr Cancer Netw. 2017;15:142–7.
Budin-Ljosne I, Teare HJ, Kaye J, Beck S, Bentzen HB, Caenazzo L, et al. Dynamic Consent: a potential solution to some of the challenges of modern biomedical research. BMC Med Ethics. 2017;18:4.
Directors Abo. Laboratory and clinical genomic data sharing is crucial to improving genetic health care: a position statement of the American College of Medical Genetics and Genomics. Genet Med. 2017;19:721–2.
Acknowledgements
We thank the patient associations BRCA France, Vaincre les Maladies Lysosomales, and APTEF Familial polyposis for their involvement. No industry or corporate sponsors contributed to the development of these recommendations. SFMPP is not funded by industrial or commercial companies but by grants from public. We thank public institutions such as the French National Cancer Institute (“Institut National du Cancer”, INCa) and, the French Foundation for Cancer Research (“Association pour la Recherche sur le Cancer”, ARC), and territorial community support (by the “Metropole of Montpellier” and the “Region Occitanie”). for donations supporting SFMPP annual meeting. Communication of the recommendations to the medical and scientific society took place in an independent framework of industrial actors, at the annual 4th SFMPP congress in Paris (June 2017).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflicts of interest with industry or firms commercializing genetic tests. No industry or corporate sponsors contributed to the development of these recommendations. SFMPP is not funded by industrial or commercial companies but by grants from public institutions and partially self-funded by SFMPP annual congresses. Dr. Pujol discloses attending Advisory Board Membership meetings for AstraZeneca, Pfizer, and Genomic Health.
Electronic supplementary material
41431_2018_224_MOESM3_ESM.png
Flowchart representing the evaluation process and ranking of the initial cancer related gene list by the medical workgroup of the SFMPP
Rights and permissions
About this article

Cite this article
Pujol, P., Vande Perre, P., Faivre, L. et al. Guidelines for reporting secondary findings of genome sequencing in cancer genes: the SFMPP recommendations. Eur J Hum Genet 26, 1732–1742 (2018). https://doi.org/10.1038/s41431-018-0224-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-018-0224-1
This article is cited by
-
Evaluation of a two-step model of opportunistic genomic screening
European Journal of Human Genetics (2024)
-
An spanish study of secondary findings in families affected with mendelian disorders: choices, prevalence and family history
European Journal of Human Genetics (2023)
-
Singleton exome sequencing of 90 fetuses with ultrasound anomalies revealing novel disease-causing variants and genotype–phenotype correlations
European Journal of Human Genetics (2022)
-
Prevalence of pathogenic germline variants in the circulating tumor DNA testing
International Journal of Clinical Oncology (2022)
-
Germline sequencing for presumed germline pathogenic variants via tumor-only comprehensive genomic profiling
International Journal of Clinical Oncology (2022)

{kind=link}