
Abstract
Approximately 2% of the human genome accounts for protein-coding genes, yet most known Mendelian disease-causing variants lie in exons or splice sites. Individuals who symptomatically present with monogenic disorders but do not possess function-altering variants in the protein-coding regions of causative genes may harbor variants in the surrounding gene regulatory domains. We present such a case: a male of Afghani descent was clinically diagnosed with Wilson Disease—a disorder of systemic copper buildup—but was found to have no function-altering coding variants in ATP7B (ENST00000242839.4), the typically causative gene. Our analysis revealed the homozygous variant chr13:g.52,586,149T>C (NC_000013.10, hg19) 676 bp into the ATP7B promoter, which disrupts a metal regulatory transcription factor 1 (MTF1) binding site and diminishes expression of ATP7B in response to copper intake, likely resulting in Wilson Disease. Our approach to identify the causative variant can be generalized to systematically discover function-altering non-coding variants underlying disease and motivates evaluation of gene regulatory variants.
Similar content being viewed by others

Introduction
Of the approximately 3 billion base pairs in the human genome, less than 2% account for protein-coding genes. Within this small fraction of genomic space are located 85–90% of validated monogenic disease-causing variants in the Human Gene Mutation Database [1]. In the case of complex diseases, however, unbiased probing suggests that a majority of contributing loci are non-coding and likely acting in cis [2, 3]. For an increasing number of Mendelian cases, causative variants have been found in gene regulatory domains as well [4]. Even so, interpretation of non-coding variants in both simple and complex diseases remains very limited.
A typical individual has over 4 million variants across the whole genome, making it a challenge to identify function-altering, non-coding variants, especially given poor understanding of how molecular function is encoded outside of genes [5]. As a strategy to simplify the challenge, we propose to examine subjects who have phenotypes of well-defined, recessive monogenic diseases but no apparent coding variants in known causative genes. For example, we recruited a 5-year-old male who was diagnosed with Wilson Disease (MIM #277900) at age 3 years and exhibited non-diagnostic results for sequencing of the causative gene ATP7B (ENST00000242839.4).
Wilson Disease is a rare, autosomal recessive Mendelian disorder that is characterized by systemic copper toxicity and is estimated to affect one in 30,000–100,000 people [6]. The disorder is caused by inactivation of the copper (Cu2+)-transporting P-type ATPase (ATP7B) [7], which typically functions to incorporate copper into apoceruloplasmin and to excrete surplus copper into the bile [8]. Disruption of ATP7B function results in pathological accumulation of copper, particularly in the liver, brain, and cornea, ultimately causing liver failure, kidney malfunction, and/or neuropsychiatric disease. ATP7B spans nearly 80 kb of the genome, in which approximately 500 Wilson Disease-causing variants have been identified [9]. Nearly all of these variants fall in coding portions or splice sites of ATP7B and typically present in compound heterozygosity in affected individuals [6, 9,10,11].
Essential heavy metals—including zinc, iron, and copper—are integral cofactors of certain enzymes and transcription factors but become extremely toxic when present in excess, as evident in Wilson Disease. Metal regulatory transcription factor 1 (MTF1) plays a key role in maintaining metal homeostasis by translocating from the cytosol to the nucleus upon heavy metal accumulation [12, 13]. Once in the nucleus, MTF1 has been shown to bind cis-acting metal response elements (5′-TGCRCNC-3′ or 3′-GNGYGCA-5′) in target gene promoters to drive the expression of metallothionein chelators, metal transporters, and other metal-inducible proteins [14, 15]. In Drosophila, MTF1 has been observed to promote expression of DmATP7 (homolog to mammalian ATP7B and ATP7A) via the 608 bp genomic region upstream of this gene in response to copper accumulation [16]. Recent empirical studies and computational analyses have generated nuanced position weight matrices (PWMs) for the MTF1 motif to allow prediction of its regulatory targets in a genome-wide manner [17]. Its role in mammalian copper homeostasis continues to be studied [18].
To test our proposed strategy for identifying non-coding, function-altering variants, we performed whole genome sequencing (WGS) of the recruited Wilson Disease patient and his unaffected mother and father. We identified one likely causative variant—a single nucleotide substitution in the promoter of ATP7B—with a segregation pattern and predicted functional effect that matches the disease pedigree. Subsequent genomic analysis and experiments support the variant as causative for Wilson Disease.
Materials and methods
Subject recruitment
The patient and unaffected parents were recruited under a protocol approved by the Stanford Institutional Review Board (IRB) for human subjects research. Written informed consent was obtained for all participants.
Whole genome sequencing
Genomic DNA from whole blood for all three subject participants was provided to Macrogen Corporation (Cambridge, MA, USA) for 35× WGS on a HiSeq X Ten (Illumina, San Diego, CA, USA). Raw sequencing reads (151 bp paired-end reads) were returned to us for detailed analysis and follow up.
Read mapping, variant calling, annotation, and filtering
Sequencing reads were mapped to the GRCh37/hg19 assembly of the human genome using the MEM algorithm of the Burrows-Wheeler Aligner, version 0.7.10-r789, with default parameters and processed as in [19]. Duplicate reads were marked with Picard Tools, version 1.105 [20]. Variants were called using the Genome Analysis Toolkit, version 3.4–46-gbc02625, following the HaplotypeCaller workflow in the Genome Analysis Toolkit Best Practices, including insertion/deletion realignment and base quality score recalibration [21]. ANNOVAR, version 527, was used to annotate variants with a predicted effect on protein-coding genes from the Ensembl gene set, version 75, and with the allele frequency observed in Exome Aggregation Consortium (ExAC) and 1000 Genomes Project (KGP) control human population [5, 22,23,24]. Variants were filtered to retain (i) only those that are rare (≤1% allele frequency in all KGP and ExAC population), and (ii) have an effect on protein-coding sequence (nonsynonymous, stopgain, stoploss, frameshift, or splicing variants, listed in Supplementary Table 1), or ones internal or within 1000 bp upstream or downstream of the canonical ATP7B isoform ENST00000242839.4, and (iii) have sufficiently many (>10) high-quality supporting reads. The likely disease-causing variant chr13:g.52,586,149T>C (NC_000013.10, hg19) has been submitted to the publicly accessible ClinVar (www.ncbi.nlm.nih.gov/clinvar/) database (ClinVarID:523102).
Binding site prediction
The tfbsPredict tool [25] was used to identify MTF1 binding sites in the ATP7B promoter. The tool was run on all k-mers within a 10 bp sequence flanking the variant of interest chr13:g.52,586,149T>C, and only predicted binding sites with a match score over 800 (out of 1000) were analyzed [25].
Cloning and genotyping
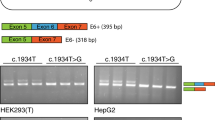
A 379 bp fragment surrounding chr13:g.52,586,149T>C was amplified from genomic DNA samples of the patient and both of his parents (Fig. 1a). The PCR reaction was performed with Q5 Hot Start High-Fidelity 2X Master Mix (New England Biolabs, Ipswich, MA, USA) using the following primers (which include homology arms for the Gibson assembly described below): 5′-ACTGGCCGGTACCTGAGCTCGCTGTGATTGACAGCCGTCGC-3′ and 5′-CAGATCTTGATATCCTCGAGGTCCGACACTGTACTGGGATCT-3′. The PCR products were then sub-cloned using the Zero Blunt TOPO PCR Cloning Kit (ThermoFisher, Waltham, MA, USA). Ten to twelve plasmid clones from each individual were subjected to Sanger sequencing (Elim Biopharmaceuticals, Hayward, CA, USA) for genotype verification (Fig. 1b). Analogous PCR amplification was performed from a separate, unrelated individual verified to be identical to the GRCh37/hg19 reference assembly in this region. The cloned fragments from the patient and this unrelated individual were inserted into the promoter-less pGL4.10 vector at the NheI site using the NEBuilder HiFi DNA Assembly Kit (New England Biolabs). As verified by Sanger sequencing, the two resulting versions of pGL4.10 differ by one nucleotide, which corresponds to chr13:g.52,586,149 T > C.

Genomic context of the candidate causative variant. a UCSC Genome Browser (hg19 minus strand) view of the region near the candidate causative variant positioned 676 bp upstream of the canonical ATP7B translation start site (chr13:g.52,586,149A>G, cyan highlight and asterisk). The variant lies in a 100-way vertebrate alignment conserved element and region of high cross-species sequence conservation as computed by PhastCons and PhyloP [30]. The chromatin surrounding chr13:g.52,586,149T>C is hypersensitive to DNaseI (and, therefore, open and accessible) in HepG2 cells. The sequence cloned into the luciferase reporter vector is indicated by the black bar. b Genotype verification for chr13:g.52,586,149T>C (red asterisk) by Sanger sequencing. Representative chromatograms show the indicated number (n=x/y) of amplification products containing the reference (T) or alternate (C) allele for each individual. Box indicates a predicted binding site for MTF1. c The candidate causative variant (bolded, pink) in the Wilson Disease (WD) patient disrupts a key base in the MTF1 position weight matrix. This exact base (boxed) is unaltered in all primates and in dozens of other mammals, even as distant as Tasmanian devil (Supplementary Fig. 1b) (color figure online).
Cell culture
HepG2 cells were cultured in RMPI 1640 medium supplemented with fetal bovine serum (10% v/v) and penicillin–streptomycin. T-125 flasks and 48-well plates were used for the ChIP-qPCR and luciferase reporter assays, respectively. Cells were passaged at 90% confluence.
Luciferase reporter assay
To assess promoter transactivation activity, the Dual-Glo® Luciferase Assay System (Promega, Madison, WI, USA) was performed according to the manufacturer’s instructions. At 70% confluence, HepG2 cells were transfected with pRL-TK control vector (15 ng per well) and pGL4.10 (200 ng per well) containing either the reference or alternate promoter allele using TransIT-X2 (Mirus Bio, Madison, WI, USA). Forty-eight hours later, the relative luciferase activity was quantified using a Berthold Technologies luminometer. For experiments involving overexpression of MTF1, the human MTF1-FLAG expression plasmid (ABclonal, Woburn, MA, USA: HG15046-CF) was transfected (200 ng per well) 24 h prior to introducing the luciferase vectors. Technical triplicates of three biological replicates were tested. Two-sample t-tests and difference-in-difference statistical analyses were performed to assess the effect significance of the alternate allele and of MTF1 concentration on ATP7B promoter activity.
ChIP-qPCR
Polyclonal rabbit anti-MTF1 antibody (Proteintech, Rosemont, IL, USA: 25383-1-AP) was used for chromatin immunoprecipitation. By comparison with anti-FLAG M2 (Sigma-Aldrich, St Louis, MO, USA: F1804), this antibody was validated for specificity by Western blotting and immunofluorescence microscopy of wildtype and MTF1-FLAG overexpressing HepG2 cells (Supplementary Figure 2). Chromatin was cross-linked for 10 min at room temperature using 0.75% formaldehyde, quenched with 0.125 M glycine, washed with ice-cold 1× PBS buffer, and sheared to 150–300 bp fragments using a Bioruptor sonicator (Diagenode Inc, Denville, NJ, USA). The ChIP was performed according to standard protocols using 0.75 µg of anti-MTF1 antibody or of non-specific isotype-matched control IgG (Life Technologies, Carlsbad, CA, USA: 026102). The immunoprecipitated DNA was reverse-crosslinked, and enrichment of loci immunoprecipitated by MTF1-specific vs. non-specific isotype-matched control antibodies was measured in technical triplicate by quantitative PCR analysis (see Supplementary Table 2 for the list of qPCR primers). A melting curve analysis was performed for each sample to ensure that a single product was amplified.
ChIP-qPCR negative control design
We obtained HepG2 open chromatin regions from the ENCODE DNase-seq dataset ENCFF373RBB. We trimmed the dataset to regions >1000 bp that have a broadPeak score of 1000 to maximize our confidence in the chromatin accessibility of the target region. From these peaks, we removed any regions with a PRISM-predicted [25] MTF1 site or with a perfect match to any of the MTF1 PWMs present in the motif library described above. From the remaining sequences, the single region with the lowest average PWM match score per nucleotide was chosen. Primers for qPCR were designed from this sequence (located in an intron of WDPCP) to which MTF1 is unlikely to bind.
Immunostaining of HepG2 cells
Cells were fixed in 4% paraformaldehyde for 10 min. Primary and secondary antibodies (diluted in 0.5% Triton X-100 in PBS) were incubated at 4 °C for at least 1 h, respectively. Each antibody incubation was followed by three 10-min PBT (PBS with 0.5% Triton X-100) washes. The following antibodies were used: anti-FLAG M2 (Sigma-Aldrich, F1804; 1:1000) and anti-MTF1 (Proteintech, 25383-1-AP; 1:1000). Secondary antibodies were Alexa Fluor conjugates (488, 555, Life Technologies; 1:250). Samples were imaged using a Zeiss Axio Observer inverted fluorescence microscope.
Results
Clinical presentation
The patient was biochemically diagnosed with Wilson Disease at age 3 years. Upon developing a high fever, he was found to have elevated liver transaminase levels (ALT = 265 U/L, AST = 124 U/L), which persisted after the fever resolved. Total and fractionated bilirubins as well as alpha-fetoprotein were normal. There was mild elevation of alkaline phosphatase at 375 U/L. Subsequent tests showed low ceruloplasmin (6 mg/dL) and serum copper (23 µg/mL) levels and abnormally high levels of copper in biopsied liver tissue (1600 µg/g dry weight). In contrast, homeostasis of other metals appeared unaffected in the patient who exhibited normal serum zinc (104 µg/dL), serum iron (75 µg/dL), and iron saturation (22%) levels. Normal reference ranges for the test results are as follows: 15–41 U/L (ALT), 3–34 U/L (AST), 104–345 U/L (alkaline phosphatase), 20–35 mg/dL (ceruloplasmin), 87–187 µg/mL (serum copper), 10–35 µg/g (copper per dry weight of liver), 56–134 µg/dL (serum zinc), 11–150 µg/dL (serum iron), 15–55% (iron saturation). At the time of diagnosis, the patient’s growth and development were typical for age. Physical examination and liver ultrasound were unremarkable with no evidence of hepatomegaly. An ophthalmology exam was performed and was normal. In addition to elevated copper levels, percutaneous liver biopsy showed subtle portal inflammation, a few necrotic hepatocytes, and focal steatosis. Currently, the patient is treated with zinc, vitamin E, and a low copper diet, and continues to meet typical growth and developmental milestones.
Gene sequencing of ATP7B was performed at the Molecular Diagnostics Laboratory of Children’s National Health System (Washington, D.C.), but no coding/splicing variants were identified. Gene deletion and duplication analyses of ATP7B were likewise negative. The subject’s parents, both of Afghani descent, and his older brother are asymptomatic and otherwise appear healthy. Given a clear clinical presentation of Wilson Disease and no detectable coding lesions, we hypothesized the presence of causative variants in the cis-regulatory domain of ATP7B.
Whole genome sequencing reveals a candidate causative variant
Following WGS of the subject trio, we considered all variants that passed variant interpretation filters (see Methods and Supplementary Table 1) and identified two candidate variants. The first single nucleotide variant (SNV), ATP7B:c.1544-634G>A (ENST00000242839.4), resides in the third intron of ATP7B (>600 bp from the nearest exon junction and >40 kb from the canonical transcription start site) and is homozygous in the patient and heterozygous in both parents. This variant is present in heterozygosity in five individuals in gnomAD [22]; many mammals exhibit the alternate allele at this locus, which does not intersect a conserved element in the UCSC 100-way vertebrate alignment (Supplementary Figure 1a); and it scores low (4.668; pathogenicity threshold > 20) in the CADD non-coding pathogenicity predictor [26]. We thus considered this variant to be likely benign.
The remaining SNV, chr13:g.52,586,149T>C (NC_000013.10, hg19), is positioned 676 bases upstream of the annotated canonical translation start site of ATP7B (Fig. 1a), in the proximal promoter. The SNV was verified by Sanger sequencing to be heterozygous in both healthy parents and homozygous in the patient (Fig. 1b). It has also been observed as heterozygous in five East Asian individuals in gnomAD [22]. Using Beagle 4.0 [27], we identified a 1,838,468 bp region of homozygosity by descent (chr13:g.51,755,329–53,593,797 [NC_000013.10, hg19]; LOD score of 77.2) in the patient that encompasses chr13:g.52,586,149T>C.
Notably, chr13:g.52,586,149T>C has previously been observed in heterozygosity in one Chinese [28] and ten Indian individuals with Wilson Disease [29]. In these instances, chr13:g.52,586,149T>C was almost always identified in conjunction with one or more heterozygous ATP7B missense variants (with exceptions where no ATP7B coding variants were detected and yet only this promoter SNV was observed as heterozygous). Additionally, chr13:g.52,586,149T>C lies in a PhastCons- and PhyloP-computed [30] region of high cross-species sequence conservation (Fig. 1a and Supplementary Figure 1b). This exact base is conserved in all primates (Fig. 1c) and in dozens of other mammals, even as distant as the marsupial Tasmanian devil, in the UCSC 100-way vertebrate alignment (Supplementary Figure 1b). Importantly, this variant would not have been identified by standard coding/splicing analysis.
MTF1 is predicted to bind at the candidate causative SNV
We searched the 10 bp conserved sequence surrounding chr13:g.52,586,149T>C for transcription factor binding motifs and found that MTF1 (ENSG00000188786.9) is predicted to bind at this locus (PWM score = 851, see Methods). Importantly, chr13:g.52,586,149T>C disrupts a key base in the MTF1 PWM (Fig. 1c), reducing the match score substantially (to 667).
Effect of chr13:g.52,586,149T>C on ATP7B promoter activity
Since hepatic copper accumulation is a key feature and contributor to the progression of Wilson Disease, we tested our hypotheses in HepG2 cells as a relevant in vitro context and model for human hepatocytes. To assess the effect of chr13:g.52,586,149T>C on the transactivation potential by the ATP7B promoter, we constructed two luciferase reporter vectors driven solely by the 379 bp sequence (Fig. 1a) surrounding the SNV locus. As verified by Sanger sequencing, these reporter vectors contain either the reference (T) or disease-associated alternate (C) allele and differ by the single nucleotide corresponding to chr13:g.52,586,149T>C. In untreated HepG2 cells, the nucleotide change resulted in a 34% decrease (Ptwo-tailed = 0.0018, Fig. 2a) in reporter expression by the alternate allele. An increase in MTF1 concentration caused a dramatic increase in transactivation from both alleles, but the effect on the alternate allele was significantly reduced compared to that on the reference allele (Ptwo-tailed = 0.0086, Fig. 2a). Analogous results were obtained with the luciferase reporter driven by a longer 986 bp sequence surrounding chr13:g.52,586,149T>C, including the core promoter and a majority of the 5′ UTR of ATP7B (data not shown).

Functional interrogation of chr13:g.52,586,149T>C. a Luciferase reporter assays in HepG2 cells were performed to quantify differences in transactivation by a 379 bp fragment of the ATP7B promoter with the chr13:g.52,586,149T (Ref) or chr13:g.52,586,149C (Alt) allele, in the presence (+) or absence (−) of MTF1 overexpression (OE). Both with and without MTF1 OE, the reference allele drove significantly higher expression compared to the alternate allele (comparison 1 and 2, respectively). Both alleles experienced dramatic increases in activity with MTF1 OE (comparisons 3 and 4), but the reference allele yielded a greater increase in expression than did the alternate allele in this context (comparison 5). Bars represent mean ± SD. Two-tailed p-values from unpaired t-tests for each comparison are shown above the associated bracket. b ChIP-qPCR was performed to determine the extent of MTF1 binding at the SNV locus (ATP7B) compared to a computationally predicted negative control (Neg Ctrl, in an intron of WDPCP) and to a previously published [32] experimentally validated locus (Pos Ctrl, in the 5′ UTR of SELENOH). Enrichment of DNA immunoprecipitated by MTF1-specific vs. non-specific isotype-matched control antibodies was measured in technical triplicate by quantitative PCR analysis. Bars represent mean ± SD. One-tailed p-values from unpaired t-tests for each comparison are shown above the associated bracket. c Proposed model for disease-causing mechanism of chr13:g.52,586,149T>C: (1) Excess intracellular accumulation of copper, Cu2+, increases (2) expression or nuclear translocation of MTF1 [12, 13]. In wildtype individuals, MTF1 then binds at chr13:g.52,586,149T (3) to recruit transcriptional machinery for upregulating ATP7B expression [34] and eliminating copper through serum ceruloplasmin and, ultimately, through the bile. We propose that the Wilson Disease (WD) patient’s homozygous single nucleotide promoter variant chr13:g.52,586,149T>C exhibits reduced affinity to MTF1. (4) The result is an insufficient ATP7B transcriptional response, consequent copper accumulation, and symptoms characteristic of Wilson Disease
ChIP binding of MTF1 to the candidate causative SNV locus
Using publicly available DNase-seq data, we verified (Fig. 1a) that the chromatin surrounding chr13:g.52,586,149T>C is open and accessible in HepG2 cells [31]. A commercially available polyclonal antibody raised against human MTF1 was validated by co-immunoprecipitation followed by SDS-PAGE analysis and by immunofluorescence microscopy for use in chromatin immunoprecipitation (ChIP, see Methods and Supplementary Figure 2). Quantitative PCR analysis of genomic DNA immunoprecipitated by endogenous MTF1 in HepG2 cells revealed a significant increase over background binding (by nonspecific, isotype-matched control IgG) to a previously validated [32] locus in the 5′ UTR of the metal-responsive gene SELENOH (NM_001321335.1, Pos Ctrl, Fig. 2b). Moreover, we indeed observed an enrichment of MTF1 binding at the SNV locus that is of similar magnitude to what was observed for the positive control site near SELENOH (Pos Ctrl, Fig. 2b). In contrast, a region that was computationally predicted to have minimal MTF1 binding (Neg Ctrl, Fig. 2b) exhibited relatively little enrichment over background binding.
Discussion
Here, we present the discovery and functional evaluation of the homozygous SNV chr13:g.52,586,149T>C in a male of Afghani descent with Wilson Disease. Based on the genomic context and inheritance pattern of chr13:g.52,586,149T>C, its disruption of a pivotal position in a predicted binding site of a copper-regulated transcription factor (MTF1), and the absence of other rare, relevantly segregating, and phenotypically and semantically congruent variants, we suggest that this SNV is the causative variant for Wilson Disease in the patient.
We provide experimental evidence that this outcome is likely explained by reduced MTF1 binding to the disease-associated promoter allele, a mechanism that has previously been suggested [33] but never directly implicated for Wilson Disease. We performed promoter reporter assays with the region immediately flanking the candidate causative variant and showed that transactivation by the disease-associated allele is significantly weaker than that by the reference allele. We further showed that transactivation by either allele of the ATP7B promoter is dramatically increased in the presence of overexpressed MTF1 protein but that the increase by the reference allele is significantly greater than that by the alternate allele. These results, in addition to enrichment of MTF1-immunoprecipitated genomic DNA at the SNV locus, strongly imply direct interaction of MTF1 with the ATP7B promoter to potentiate transcription.
Based on these findings, we suggest a model of the disease-causing mechanism as follows (Fig. 2c): Excess intracellular accumulation of copper increases nuclear translocation of MTF1 [12, 13]. In healthy individuals, MTF1 then binds at chr13:g.52,586,149T to recruit transcriptional machinery to upregulate ATP7B expression [34] and eliminate copper through serum ceruloplasmin and bile. We propose that the patient’s homozygous SNV, chr13:g.52,586,149T>C, exhibits reduced affinity to MTF1. The result is an insufficient ATP7B transcriptional response, consequent copper accumulation, and symptoms characteristic of Wilson Disease.
There exist many individuals who are heterozygous (carriers) for deleterious nonsense or structural variants of ATP7B and yet do not exhibit symptoms of Wilson Disease [11]. One interpretation of this scenario is that 50% of wildtype ATP7B concentration is sufficient for maintaining copper homeostasis; in contrast, the promoter variant chr13:g.52,586,149T>C appears to reduce expression of ATP7B by only 34% and yet still results in deranged copper metabolism. This seemingly incongruent conclusion can be reconciled by considering effects of genetic compensation and trans-acting feedback. If levels of functional ATP7B protein are insufficient, ATP7B mRNA transcript levels may be upregulated—through binding of transcription factors at regulatory loci, likely including chr13:g.52,586,149T>C—to maintain adequate protein concentration. If this were the case, heterozygous carriers of ATP7B loss-of-function variants would be able to achieve protein levels higher than 50% of wildtype through the remaining unaffected allele. In this study’s patient, however, the cis-regulatory apparatus for ATP7B appears to be inherently defective for both alleles (since the SNV is homozygous), precluding sufficient compensation that a single wildtype allele with functional regulatory input could otherwise readily provide.
A handful of other disease-associated variants in the ATP7B promoter of Wilson Disease subjects has been observed but almost always in compound heterozygosity with missense variants—with exceptions where the additional variant has not yet been detected [28, 29, 35,36,37]. The one previously reported instance of a homozygous ATP7B promoter variant consists of a 15 bp deletion upstream of the canonical translation start site (chr13:g.52,585,895_52,585,909del [NC_000013.10, hg19], which corresponds to the deletion 441 to 427 bp upstream of the translation start site in the isoform described in Loudianos et al. [35]). This deletion is predicted to disrupt a FOXA family transcription factor binding site (though no direct functional validation has been performed) and is exclusively seen in subjects of Sardinian descent, possibly arising from a founder effect [35, 37].
To our knowledge, the present finding is the first reported instance of a homozygous, cis-acting SNV in the ATP7B promoter to cause Wilson Disease and the first time MTF1 has been directly implicated in this context. Despite the difference of just one putatively causative nucleotide, the patient exhibited abnormal copper levels equivalent to those of individuals with biallelic coding variants in ATP7B [38]. Our experimental evaluation of this single nucleotide promoter variant also adds to the growing list of Mendelian disease presentations that are caused by cis-regulatory SNVs disrupting the binding of key transcription factors [39, 40].
This study illustrates a promising example of identifying novel disease-causing cis-regulatory variants and the regulatory factor being disrupted by examining individuals who have well-characterized recessive, monogenic disorders but do not exhibit coding/splicing variants in the known causative gene. Because our strategy leverages preexisting knowledge of the disease-associated gene, the search space and interpretation of otherwise hard-to-decipher, non-coding variants becomes much more tractable. Likewise, the number of candidate variants is also minimized when considering recessive disorders that necessarily derive from homozygous or compound heterozygous lesions in a single gene (or its regulatory domain). Our findings also indicate that, until WGS becomes ubiquitous, expansion of target gene sequencing to include at least 1 kb of the proximal promoter would be useful for improving the rate of diagnosis for Mendelian disease cases.
References
Stenson PD, Ball EV, Mort M, Phillips AD, Shiel JA, Thomas NST, et al. Human Gene Mutation Database (HGMD): 2003 update. Hum Mutat. 2003;21:577–81.
Zhang F, Lupski JR. Non-coding genetic variants in human disease. Hum Mol Genet. 2015;24:R102–10.
Petit F, Sears KE, Ahituv N. Limb development: a paradigm of gene regulation. Nat Rev Genet. 2017;18:245–58.
Spitz F. Gene regulation at a distance: from remote enhancers to 3D regulatory ensembles. Semin Cell Dev Biol. 2016;57:57–67.
Auton A, Abecasis GR, Altshuler DM, Durbin RM, Bentley DR, Chakravarti A, et al. A global reference for human genetic variation. Nature. 2015;526:68–74.
Ala A, Walker AP, Ashkan K, Dooley JS, Schilsky ML. Wilson’s disease. Lancet. 2007;369:397–408.
Petrukhin K, Lutsenko S, Chernov I, Ross BM, Kaplan JH, Gilliam TC. Characterization of the Wilson disease gene encoding a P-type copper transporting ATPase: genomic organization, alternative splicing, and structure/function predictions. Hum Mol Genet. 1994;3:1647–56.
de Bie P, Muller P, Wijmenga C, Klomp LWJ. Molecular pathogenesis of Wilson and Menkes disease: correlation of mutations with molecular defects and disease phenotypes. J Med Genet. 2007;44:673–88.
Bandmann O, Weiss KH, Kaler SG. Wilson’s disease and other neurological copper disorders. Lancet Neurol. 2015;14:103–13.
Todorov T, Balakrishnan P, Savov A, Socha P, Schmidt HHJ. Intragenic deletions in ATP7B as an unusual molecular genetics mechanism of Wilson’s disease pathogenesis. PLoS ONE. 2016;11:1–11.
Coffey AJ, Durkie M, Hague S, McLay K, Emmerson J, Lo C, et al. A genetic study of Wilson’s disease in the United Kingdom. Brain. 2013;136:1476–87.
Vanacore RM, Eskew JD, Morales PJ, Sung L, Smith A. Role for copper in transient oxidation and nuclear translocation of MTF-1, but not of NF-κB, by the heme–hemopexin transport system. Antioxid Redox Signal. 2000;2:739–52.
Saydam N, Georgiev O, Nakano MY, Greber UF, Schaffner W. Nucleo-cytoplasmic trafficking of metal-regulatory transcription factor 1 is regulated by diverse stress signals. J Biol Chem. 2001;276:25487–95.
Labbé S1, Prévost J, Remondelli P, Leone A, Séguin C. A nuclear factor binds to the metal regulatory elements of the mouse gene encoding metallothionein-l. Nucleic Acids Res. 1991;19:4225–31.
Larochelle O, Stewart G, Moffatt P, Tremblay V, Séguin C. Characterization of the mouse metal-regulatory-element-binding proteins, metal element protein-1 and metal regulatory transcription factor-1. Biochem J. 2001;353:591–601.
Burke R, Commons E, Camakaris J. Expression and localisation of the essential copper transporter DmATP7 in Drosophila neuronal and intestinal tissues. Int J Biochem Cell Biol. 2008;40:1850–60.
Wang M, Yang F, Zhang X, Zhao H, Wang Q, Pan Y. Comparative analysis of MTF-1 binding sites between human and mouse. Mamm Genome. 2010;21:287–98.
Grzywacz A, Gdula-Argasińska J, Muszyńska B, Tyszka-Czochara M, Librowski T, Opoka W. Metal responsive transcription factor 1 (MTF-1) regulates zinc dependent cellular processes at the molecular level. Acta Biochim Pol. 2015;62:491–8.
Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. 2013. http://arxiv.org/abs/1303.3997.
Broad Institute. Picard Tools.
DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43:491–8.
Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–91.
Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164.
Yates A, Akanni W, Ridwan MR, Barrell D, Billis K, Carvalho-Silva D, et al. Ensembl 2016. Nucleic Acids Res. 2016;44:D710–6.
Wenger AM, Clarke SL, Guturu H, Chen J, Schaar BT, McLean CY, et al. PRISM offers a comprehensive genomic approach to transcription factor function prediction. Genome Res. 2013;23:889–904.
Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46:310–5.
Browning BL, Browning SR. Improving the accuracy and efficiency of identity-by-descent detection in population data. Genetics. 2013;194:459–71.
Lu C-X, Lin Q, Huang W-Q, Tzeng C-M. New mutations and polymorphisms of the ATP7B gene in sporadic Wilson disease. Eur J Med Genet. 2014;57:498–502.
Mukherjee S, Dutta S, Majumdar S, Biswas T, Jaiswal P, Sengupta M, et al. Genetic defects in Indian Wilson disease patients and genotype–phenotype correlation. Parkinsonism Relat Disord. 2014;20:75–81.
Siepel A, Bejerano G, Pedersen JS, Hinrichs AS, Hou M, Rosenbloom K, et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005;15:1034–50.
ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74.
Stoytcheva ZR, Vladimirov V, Douet V, Stoychev I, Berry MJ. Metal transcription factor-1 regulation via MREs in the transcribed regions of selenoprotein H and other metal-responsive genes. Biochim Biophys Acta. 2010;1800:416–24.
Günther V, Lindert U, Schaffner W. The taste of heavy metals: gene regulation by MTF-1. Biochim Biophys Acta. 2012;1823:1416–25.
Hardyman JE, Tyson J, Jackson KA, Aldridge C, Cockell SJ, Wakeling LA, et al. Zinc sensing by metalresponsive transcription factor 1 (MTF1) controls metallothionein and ZnT1 expression to buffer the sensitivity of the transcriptome response to zinc. Metallomics. 2016;8:337–43.
Loudianos G, Dessi V, Lovicu M, Angius A, Figus A, Lilliu F, et al. Molecular characterization of wilson disease in the Sardinian population—evidence of a founder effect. Hum Mutat. 1999;14:294–303.
Wan L, Tsai CH, Hsu CM, Huang CC, Yang CC, Liao CC, et al. Mutation analysis and characterization of alternative splice variants of the Wilson disease gene ATP7B. Hepatology. 2010;52:1662–70.
Cullen LM, Prat L, Cox DW. Genetic variation in the promoter and 5′ UTR of the copper transporter, ATP7B, in patients with Wilson disease. Clin Genet. 2003;64:429–32.
Shah AB, Chernov I, Zhang HT, Ross BM, Das K, Lutsenko S, et al. Identification and analysis of mutations in the Wilson disease gene (ATP7B): population frequencies, genotype–phenotype correlation, and functional analyses. Am J Hum Genet. 1997;61:317–28.
de Vooght KMK, van Wijk R, van Solinge WW. Management of gene promoter mutations in molecular diagnostics. Clin Chem. 2009;55:698–708.
Corradin O, Scacheri PC. Enhancer variants: evaluating functions in common disease. Genome Med. 2014;6:85.
Acknowledgements
We thank the subject family for consenting to this study; Eric Rulifson for key technical guidance on the luciferase reporter assays; Ashby Morrison, Kristy Red-Horse, and Roel Nusse for lending experimental equipment; Alan Boyle, Lingyun Song, and Greg Crawford from Duke University’s Institute for Genome Sciences & Policy (IGSP) for generating the publicly available HepG2 DNase-seq data; and members of the Bejerano Lab for helpful discussions, suggestions, and support.
Funding
This study was funded in part by the National Science Foundation Graduate Research Fellowship to HIC, the Stanford Graduate Fellowship and the Computational, Evolutionary and Human Genetics Fellowship to KAJ, the Bio-X Stanford Interdisciplinary Graduate Fellowship to JB, the Stanford Pediatrics Department, DARPA, a Packard Foundation Fellowship and a Microsoft Faculty Fellowship to GB.
Author contributions
SS recruited and consented the study subjects. KAJ, JB, AMW, HG, and GB designed the computational approach. HIC designed and performed the experimental evaluation. JB assisted with experiments. KAJ performed the computational analysis. HIC wrote the manuscript with input from all authors. JAB and GB supervised the project.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Chen, H.I., Jagadeesh, K.A., Birgmeier, J. et al. An MTF1 binding site disrupted by a homozygous variant in the promoter of ATP7B likely causes Wilson Disease. Eur J Hum Genet 26, 1810–1818 (2018). https://doi.org/10.1038/s41431-018-0221-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-018-0221-4
This article is cited by
-
Missing heritability of Wilson disease: a search for the uncharacterized mutations
Mammalian Genome (2023)
-
Investigation of the Wilson gene ATP7B transcriptional start site and the effect of core promoter alterations
Scientific Reports (2021)
