
Abstract
We have developed and validated for the diagnosis of inherited colorectal cancer (CRC) a massive parallel sequencing strategy based on: (i) fast capture of exonic and intronic sequences from ten genes involved in Mendelian forms of CRC (MLH1, MSH2, MSH6, PMS2, APC, MUTYH, STK11, SMAD4, BMPR1A and PTEN); (ii) sequencing on MiSeq and NextSeq 500 Illumina platforms; (iii) a bioinformatic pipeline that includes BWA-Picard-GATK (Broad Institute) and CASAVA (Illumina) in parallel for mapping and variant calling, Alamut Batch (Interactive BioSoftware) for annotation, CANOES for CNV detection and finally, chimeric reads analysis for the detection of other types of structural variants (SVs). Analysis of 1644 new index cases allowed the identification of 323 patients with class 4 or 5 variants, corresponding to a 20% disease-causing variant detection rate. This rate reached 37% in patients with Lynch syndrome, suspected on the basis of tumour analyses. Thanks to this strategy, we detected overlapping phenotypes (e.g., MUTYH biallelic mutations mimicking Lynch syndrome), mosaic alterations and complex SVs such as a genomic deletion involving the last BMPR1A exons and PTEN, an Alu insertion within MSH2 exon 8 and a mosaic deletion of STK11 exons 3–10. This strategy allows, in a single step, detection of all types of CRC gene alterations including SVs and provides a high disease-causing variant detection rate, thus optimizing the diagnosis of inherited CRC.
Similar content being viewed by others


Introduction
Identification of a disease-causing variant involved in Mendelian forms of colorectal cancer (CRC) is essential to ensure, in carriers, an early detection of colorectal tumours using chromocolonoscopy and to suppress, in non-carriers, an inappropriate medical follow-up and illegitimate anxiety. The Mendelian forms of CRC include: (i) Lynch syndrome, resulting from heterozygous variants of the MMR genes, MSH2 (MIM *609309), MSH6 (MIM *600678), MLH1 (MIM *120436) and PMS2 (MIM *600259); (ii) autosomal-dominant and -recessive adenomatous polyposis due, respectively, to heterozygous variants of APC (MIM *611731) and biallelic MUTYH (MIM *604933) variants and (iii) autosomal-dominant hamartomatous polyposis including juvenile polyposis due to SMAD4 (MIM* 600993) or BMPR1A (MIM* 601299) variants, Peutz–Jeghers syndrome linked to STK11 (MIM* 602216) variants and Cowden syndrome resulting from PTEN (MIM* 601728) alterations (for review see, references [1, 2]). In patients or families strongly suspected to present an inherited form of CRC, but without detectable alterations within these genes, studies based on whole-exome sequencing have recently allowed the identification, in a limited number of families, of rare disease-causing variants within other genes: POLE (MIM* 174762), POLE2 (MIM* 602670) and POLD1 (MIM* 174761) encoding the subunits of polymerase epsilon or delta enzyme complexes and which germline alterations define the autosomal-dominant polymerase-proofreading-associated polyposis [3]; the base-excision repair genes NTHL1 (MIM* 602656) [4] and MSH3 (MIM* 600887) [5], both being involved in rare autosomal recessive polyposes. Nevertheless, the cancer risks associated with the variants detected within these genes remain to be characterized.
We report here our experience on 1761 patients, including 1644 new patients, of the optimization of inherited CRC diagnosis, using NGS of a gene panel. Our guidelines were: (i) to only include in the panel those genes whose involvement in Mendelian forms of CRC had been validated, (ii) to capture exonic and intronic regions in order to facilitate the detection of copy number variations (CNVs) and other structural variants (SVs), (iii) to sequence at a sufficient depth to facilitate mosaic alteration detection and (iv) to integrate quality control procedures at each step of the diagnostic process.
Patients and methods
All patients included in this study have been observed in the context of a genetic session and were analyzed because their clinical presentation was suggestive of an inherited CRC (Table 1): MMR-deficient tumours <65 years; familial aggregation of CRC including one case <51 years; early-onset CRC (<41 years); adenomatous polyposis (as defined by more than ten histologically proven adenomas); one advanced adenoma (adenoma ≥10 mm, tubulovillous, high-grade dysplasia); several adenomas (2–10) <61 years; hamartomatous polyposis (as defined by the presence of histologically proven hamartomas); or serrated polyposis syndrome (as defined by the presence of more than five serrated lesions). All patients had given their informed consent for genetic analysis. Two consecutive series of patients, corresponding to 70 patients harbouring known alterations (70 SNVs, 31 Indels and 12 CNVs) within the CRC genes, and 47 patients analyzed in parallel using conventional procedures based on Sanger sequencing and QMPSF were used to validate the process and bioinformatic pipelines.
DNA was extracted from peripheral blood using the QuickGene-610L platform (Kurabo Biomedical, FujiFilm). Gene panel was designed using Agilent SureDesign (Agilent, Santa Clara, California) to capture the exonic and intronic sequences plus 15 kb of 5′ and 3′ sequences of the following genes: MLH1, MSH2, MSH6, PMS2 (exons 6–15), APC, MUTYH, STK11, SMAD4, BMPR1A and PTEN. The total captured sequences represented 488 kb. We set up a fast library preparation protocol using the QXT SureSelect enrichment kit from Agilent. Genomic DNA (50 ng) was tagmented using a transposase, and 10 or 80 independent libraries were then pooled after the enrichment step. This protocol was automated on a Sciclone NGSx workstation (PerkinElmer, Waltham, Massachusetts, USA) and the entire library preparation process could be achieved within 2 working days. Libraries were sequenced on a MiSeq or a NextSeq 500 platform (Illumina, San Diego, USA) using 2× 150 bp paired-end sequencing. The panel design and the enrichment protocol are available upon request.
A double bioinformatic pipeline was set up to secure detection of SNVs and Indels. The first pipeline included the CASAVA suite v1.8.2 from Illumina for demultiplexing, generation of fastq files, mapping of the reads and variant calling. In the second pipeline, reads mapping was performed using BWA, recalibration and variant calling were performed using Picard and GATK software (Broad Institute). Variants detected by one or both these pipelines were then annotated by Alamut Batch (Interactive BioSoftware, Rouen, France). Detection of CNVs was achieved using the CANOES software [6]. Chimeric reads were searched in patients whose clinical presentation was strongly suggestive of a deleterious alteration, but in whom no alteration could be detected after this first analysis. For each sequencing run, PDF quality reports integrating the number of clusters/mm2, percentage of bases with a Qscore >30, FastQC reports, percentage of mapped reads, on- and off-targets percentages, percentage of covered bases and mean sequencing depth were automatically generated using the home made tool PyQua (Python Qualitics). Only biallelic MUTYH variants were considered as disease-causing variants. Detected variants were confirmed on an independent blood sample by PCR-amplification of genomic fragments, Sanger sequencing or QMPSF (quantitative multiplex PCR of short fluorescent fragments), or MLPA (MRC-Holland). Variants and phenotype data were submitted to the InSiGHT public database (https://www.insight-database.org/variants; variant IDs: 27544–27877). For patients without detectable alteration, identity of the sample was systematically controlled on an independent blood sample using a multiplex SNaPshot analysis comparing five SNPs located within the captured regions: MLH1 NG_007109.2(NM_000249.3):c.1668–19A>G (0,4442); APC (NM_000038.5):c.1458T>C (0,5910); MSH6 (NM_000179.2):c.540T>C (0,2981); PTEN NG_007466.2(NM_000314.6):c.1026+32T>G (0,3333) and BMPR1A (NM_004329.2):c.4C>A (0,2576) (numbers in brackets indicate minor allelic frequency in Non-Finnish European populations, according to gnomAD database). Exons are numbered according to the given reference.
Results and discussion
Our captured design and NGS workflow ensured coverage of more than 99% of targeted bases and a mean read depth of 600×. All the alterations detected by Sanger sequencing and QMPSF were successfully identified in our validation series composed of 117 patients. One variant of MSH2 NG_007110.2 (NM_000251.2:c.942+3A>T), located within a poly-A stretch and near polymorphic deletion, was only detected by the CASAVA pipeline. Two Indels, located within MSH6 and STK11, were detected only by the BWA-GATK pipeline, because of a low read depth caused by a high GC content and small tandem duplication, respectively. The 12 CNVs, previously detected using QMPSF, were correctly identified using CANOES. Our NGS workflow was then applied to 1644 new patients suspected to present an inherited form of CRC. We identified 323 patients with class 4 or 5 variants (Table 1), whose pathogenicity was established according to the InSiGHT criteria for the MMR genes [7] and to the American College of Medical Genetics criteria [8] for the other genes. This corresponds to a 20% disease-causing variant detection rate. It should be noticed that the distribution of the variants presented in Table 1 does not reflect the general contribution of the corresponding variations to CRC, since our series were biased by tumour analyses. Disruptive (non-sense, frameshift and canonical splice site variations), splicing, missense variations or in-frame Indels and SVs represented 58, 7, 20 and 15% of the 344 detected disease-causing alterations including 21 biallelic MUTYH variants. The disease-causing variant detection rate reached 37% in patients with Lynch syndrome, suspected on the basis of tumour analyses. We also identified 141 class 3 variants (MSH2: 27; MLH1: 23; MSH6: 34; PMS2: 3; APC: 32; MUTYH: 4 (biallelic); BMPR1A: 5; SMAD4: 4; PTEN: 1 and STK11: 4).
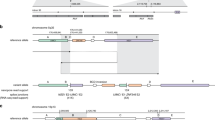
As compared to conventional methods, this strategy based on the capture of exonic and intronic sequences and NGS of a gene panel not only increases analysis throughput and reduces result delay, but has also, as illustrated by the following case reports, three main additional advantages. (i) The first is the detection of overlapping phenotypes: in a female patient presenting at 41 years of age a caecal adenocarcinoma and four metachronous adenomas without familial history of CRC, the presence of a MSI phenotype associated with loss of the MLH1/PMS2 dimer expression in the tumour led us to initially consider the diagnosis of Lynch syndrome. However, no deleterious alteration was detected within MLH1 or PMS2, but simultaneous analysis of the MUTYH revealed a common biallelic alteration (NM_001048171.1: c.[494A>G];[1145G>A]; p.[Tyr165Cys];[Gly382Asp]), revealing that, as previously described, germline inactivation of a base-excision repair gene can result into somatic alteration of the MMR genes and mimic Lynch syndrome [9, 10]. (ii) The second advantage is the detection of mosaic alterations as illustrated in Fig. 1. We detected two cases of mosaic APC variants revealed by 18 and 9% of the reads in two patients presenting, respectively, an attenuated form of adenomatous polyposis at 52 years and a diffuse form at 38 years. Furthermore, we identified three disease-causing variations of STK11 in mosaic state in three independent Peutz–Jeghers patients (revealed by 8, 28 and 38% of reads, respectively). Nevertheless, the mean depth (600×) ensured by our workflow does not guarantee the detection of all mosaics, which would require to analyze colorectal tissues [11]. (iii) The third advantage is the detection of complex SVs and accurate characterization of their breakpoints. In a female patient presenting with a mixed juvenile and adenomatous polyposis at 21 years of age, we detected a large genomic deletion (hg19 chr10:g.88678073_89742644del), removing the last exons of the BMPR1A gene and the entire PTEN gene, corresponding therefore to the 10q gene deletion syndrome previously described [12]. In a patient who developed two metachronous CRC (28 and 51 years) with a MSI phenotype and a loss of MSH2-MSH6 expression, then a duodenum carcinoma at 56 years, we detected the insertion of an Alu element at the end of exon 8 of MSH2 (Fig. 2). More remarkably, we detected a complex mosaic SV in a female patient of 28 years of age presenting with a typical and severe form of Peutz–Jeghers syndrome, but without alterations detected by conventional procedures; this patient, with mucocutaneous pigmentation of the lips, underwent at 4 months of age a left hepatectomy for several liver hamartomas and presented, since 5 years of age, recurrent upper intestinal intussusceptions and obstructions due to hamartomatous polyps that led to multiple polypectomies and segmentary resections of the small bowel and right colon and, since 22 years of age she underwent the resection of ten uterine hamartomas. This typical presentation prompted us to re-analyze STK11 using our NGS workflow. Visual screening of chimeric reads from the BAM files allowed us to detect a mosaic deletion of STK11 removing exons 3–10 and extending to exon 4 of the adjacent CBARP gene (Fig. 3a), and to accurately map the breakpoints of this 16.3 kb deletion (hg19 chr19:g.1219177_1235441del). We designed a specific PCR-amplification for the rearranged allele, which confirmed the deletion (Fig. 3b, c), and which allowed us to offer a prenatal diagnosis to this patient. As shown in Fig. 3d, this mosaic SV was hardy detectable by MLPA.

BAM visualization, using Alamut visual software (Interactive Biosoftware), of the mosaic APC variants identified in two patients. Partial sequence of APC NM_000038.5 is shown. The arrows indicate in a and b, respectively, the c.4068del, p.(Gly1357Glufs*58) and c.497dup, p.(Lys167*) mosaic APC variants. Paired-end DNA sequencing reads are visualized in blue (strands +) and green (strands −). The total number of reads, the percentage and the number of reads on each strand are indicated for the wild-type and the mutant alleles

Detection of an Alu insertion within MSH2. a BAM visualization, using Alamut visual software (Interactive Biosoftware), of the Alu element insertion in exon 8 of MSH2. Partial sequence of MSH2 NG_007110.2(NM_000251.2) is shown. Chimeric reads, corresponding to MSH2 exon 8 followed by AluY sequence, are considered as excluded bases in the BAM alignment process. b BAM visualization, in a control subject, of the same MSH2 region, showing the absence of chimeric reads

Detection of a mosaic STK11 deletion. a BAM visualization, using Integrative Genomics Viewer, of the deletion of STK11 removing exons 3–10 NG_007460.2(NM_000455.4) and extending to exon 4 of the adjacent CBARP gene NC_000019.10(NM_152769.2). Red segments represent the 16.3 kb deletion. Arrows indicate the genomic orientation of the genes. b Gel electrophoresis of the multiplex PCR-amplified wild-type (355 bp) and rearranged (209 bp) STK11 alleles in a control individual (C) and the patient (P). M designs the molecular weight marker. c Sanger sequencing of the rearranged STK11 allele. The box corresponds to the overlapping STK11 and CBARP sequences. d MLPA analysis of STK11 in a control individual (C1), a positive control with a heterozygous deletion of STK11 exons 2–10 (C2) and in the patient with the mosaic STK11 exons 3–10 deletion (P)
In conclusion, our NGS workflow based on the capture of exonic and intronic sequences, and a double pipeline with complementary algorithms allows a high disease-causing variant detection rate and, in a single step, the detection of the different types of gene alterations including SVs of the different CRC genes, and therefore a reduction of result delay for the patient benefit. Several recent reports performed on smaller series and not based on capture of exonic and intronic sequences have already shown the efficiency of NGS in the diagnosis of inherited CRC, but the reported disease-causing variant detection rate was lower or the detected variants did not include the mosaic alterations or complex SVs [13,14,15,16]. The panel that we used in this study can easily be extended to other genes, and we recently added to the panel POLE, POLD1, NTHL1 and MSH3 genes. Nevertheless, as recently highlighted, it is crucial to carefully evaluate the clinical relevance of proposed cancer susceptibility genes before their inclusion into diagnosis panels [17]. The increase of sequencing depth and additional implementation of new algorithms focused on more complex rearrangements, such as inversions, should in the future also increase the sensitivity of this strategy. In the present context of the exponential use of enlarged cancer gene panels, which exposes to an increased detection of variants of unknown significance, we think that the strategy described in this report is, for the molecular diagnosis of inherited CRC, efficient, medically relevant and cost-effective.
References
Vasen H, Tomlinson I, Castells A. Clinical management of hereditary colorectal cancer syndromes. Nat Rev Gastroenterol Hepatol. 2015;12:88–97.
Kanth P, Grimmett J, Champine M, Burt R, Samadder NJ. Hereditary colorectal polyposis and cancer syndromes: a primer on diagnosis and management. Am J Gastroenterol. 2017;112:1509–25.
Palles C, Cazier JB, Howarth KM, et al. Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nat Genet. 2013;45:136–44.
Weren RD, Ligtenberg MJ, Kets CM, et al. A germline homozygous mutation in the base-excision repair gene NTHL1 causes adenomatous polyposis and colorectal cancer. Nat Genet. 2015;47:668–71.
Adam R, Spier I, Zhao B, et al. Exome sequencing identifies biallelic MSH3 germline mutations as a recessive subtype of colorectal adenomatous polyposis. Am J Hum Genet. 2016;99:337–51.
Backenroth D, Homsy J, Murillo LR, et al. CANOES: detecting rare copy number variants from whole exome sequencing data. Nucleic Acids Res. 2014;42:e97.
Thompson BA, Spurdle AB, Plazzer JP, et al. Application of a 5-tiered scheme for standardized classification of 2,360 unique mismatch repair gene variants in the InSiGHT locus-specific database. Nat Genet. 2014;46:107–15.
Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequencevariants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24.
Morak M, Heidenreich B, Keller G, et al. Biallelic MUTYH mutations can mimic Lynch syndrome. Eur J Hum Genet. 2014;22:1334–7.
Castillejo A, Vargas G, Castillejo MI, et al. Prevalence of germline MUTYH mutations among Lynch-like syndrome patients. Eur J Cancer. 2014;50:2241–50.
Spier I, Drichel D, Kerick M, et al. Low-level APC mutational mosaicism is the underlying cause in a substantial fraction of unexplained colorectal adenomatous polyposis cases. J Med Genet. 2016;53:172–9.
Delnatte C, Sanlaville D, Mougenot JF, et al. Contiguous gene deletion within chromosome arm 10q is associated with juvenile polyposis of infancy, reflecting cooperation between the BMPR1A and PTEN tumor-suppressor genes. Am J Hum Genet. 2006;78:1066–74.
Rey JM, Ducros V, Pujol P, et al. Improving mutation screening in patients with colorectal cancer predisposition using next-generation sequencing. Clin Genet. 2017;92:405–14.
Hansen MF, Johansen J, Sylvander AE, et al. Use of multigene-panel identifies pathogenic variants in several CRC-predisposing genes in patients previously tested for Lynch syndrome. J Mol Diagn. 2017;19:589–601.
Soares BL, Brant AC, Gomes R, et al. Screening for germline mutations in mismatch repair genes in patients with Lynch syndrome by next generation sequencing. Fam Cancer. 2017. https://doi.org/10.1007/s10689-017-0043-5.
Stoffel EM, Koeppe E, Everett J, et al. Germline genetic features of young individuals with colorectal cancer. Gastroenterology. 2017. https://doi.org/10.1053/j.gastro.2017.11.004.
Broderick P, Dobbins S, Chubb D, et al. Validation of recently proposed colorectal cancer susceptibility gene variants in an analysis of families and patients—a systematic review. Gastroenterology. 2017;152:75–7.
Acknowledgements
The authors thank all the clinical and molecular geneticists who referred the patients. This programme was founded by the French Institut National du Cancer (INCa) and co-supported by European Union and Région Normandie (European Regional Development Fund, ERDF).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Baert-Desurmont, S., Coutant, S., Charbonnier, F. et al. Optimization of the diagnosis of inherited colorectal cancer using NGS and capture of exonic and intronic sequences of panel genes. Eur J Hum Genet 26, 1597–1602 (2018). https://doi.org/10.1038/s41431-018-0207-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-018-0207-2
This article is cited by
-
Detection of a pathogenic Alu element insertion in PALB2 gene from targeted NGS diagnostic data
European Journal of Human Genetics (2022)
-
Detecting inversions in routine molecular diagnosis in MMR genes
Familial Cancer (2022)
-
SVA retrotransposon insertion in exon of MMR genes results in aberrant RNA splicing and causes Lynch syndrome
European Journal of Human Genetics (2021)
-
Detection of copy-number variations from NGS data using read depth information: a diagnostic performance evaluation
European Journal of Human Genetics (2021)
