
Abstract
RASA1-related disorders are vascular malformation syndromes characterized by hereditary capillary malformations (CM) with or without arteriovenous malformations (AVM), arteriovenous fistulas (AVF), or Parkes Weber syndrome. The number of cases reported is relatively small; and while the main clinical features are CMs and AVMs/AVFs, the broader phenotypic spectrum caused by variants in the RASA1 gene is still being defined. Here, we report the clinical and molecular findings in 69 unrelated cases with a RASA1 variant identified at ARUP Laboratories. Sanger sequencing and multiplex ligation-dependent probe amplification were primarily used to evaluate RASA1. Several atypical cases were evaluated using next-generation sequencing (NGS) and array-comparative genomic hybridization (aCGH). Sixty individuals had a deleterious RASA1 variant of which 29 were novel. Nine individuals had a variant of uncertain significance. Five large RASA1 deletions were detected, giving an overall deletion/duplication rate of 8.3% (5/60) among positive cases. Most (75.4%) individuals with a RASA1 variant had CMs, and 44.9% had an AVM/AVF. Clinical findings in several cases expand the RASA1 phenotype. Our data suggest that screening for large RASA1 deletions and duplications in this disorder is important and suggest that NGS multi-gene panel testing is beneficial for the molecular diagnosis of cases with complex vascular phenotypes.
Similar content being viewed by others

Introduction
Capillary malformation-arteriovenous malformation syndrome (CM-AVM) (OMIM #608354) is a recently recognized autosomal dominant inherited disorder that occurs in 1:100,000 individuals [1]. CM-AVM is caused by mutations in RASA1 which encodes the Ras p21 protein activator 1. RASA1 is a member of the GAP1 family of GTPase-activating proteins, and is involved in regulating cell differentiation and proliferation, likely during angiogenesis [2, 3].
Early reports of CM-AVM/RASA1-related disorders emphasized the hallmark small, multiple cutaneous capillary malformations (CM) that can be found randomly distributed across the body. The CMs that typify this disorder are <1–2 cm, round to oval, pink-to-red macules, some surrounded by a pale halo [2, 3]. Other reports have expanded the description of cutaneous vascular lesions observed in patients with the disorder to include larger (5–22 cm), more irregularly shaped CMs; as well as pinpoint sized red lesions with a halo [4, 5]. The distinct white halos surrounding the telangiectases are often more prominent than the tiny punctate pink lesion in the middle [5, 6].
Approximately 30% of affected individuals also have fast-flow AVM or arteriovenous fistulas (AVF) [1, 4]. AVMs and AVFs have been identified in the brain, skin, muscle, bone, and spine in patients with a RASA1 mutation [1, 7]. Life-threatening complications can arise from these fast-flow lesions including hemorrhage, neurological consequences, congestive heart failure, and cutaneous ischemia requiring transarterial embolization or limb amputation [8]. Recently, the prenatal diagnosis of cerebral and extra-cerebral high-flow lesions was reported in two unrelated cases leading to the diagnosis of a previously undiagnosed CM-AVM syndrome in each family caused by RASA1 mutations [9].
RASA1 variants have been found in individuals diagnosed with Parkes Weber syndrome (PKWS) (OMIM #608355). PKWS is characterized by multiple micro-AVFs associated with a cutaneous capillary stain and excessive soft tissue and skeletal growth of an affected limb [1]. In one study, a RASA1 mutation was identified in 13 of 16 (81%) probands who had the phenotype of PKWS with multifocal CMs [1]. Recently, lymphatic manifestations such as lymphedema and chylothorax were described as part of the phenotypic spectrum of RASA1-related disorders [10]. RASA1 variants have additionally been reported in individuals clinically suspected to have hereditary hemorrhagic telangiectasia (HHT) who had epistaxis and dermal lesions described as telangiectases [11].
Molecular testing for RASA1 variants is useful in individuals with multifocal CMs [12]. RASA1 mutation penetrance is high (>95%) and de novo occurrence is around 30% [1]. The identification of a RASA1 variant provides indication to screen for internal AVMs or AVFs, which are generally detectable using magnetic resonance imaging (MRI) of the brain and spine. In 2012, we reported a 29% mutation detection rate among all samples sent for RASA1 clinical molecular testing (10/35) which increased to ~39% (10/26) if patients without CMs were excluded [12]. Recently, somatic second hit RASA1 variants were identified in tissue samples from capillary malformations indicating that a loss of function of both RASA1 alleles facilitates development of vascular lesions in CM-AVM patients [10, 13].
To date, the number of cases reported with a RASA1 variant is still relatively small; and while the main clinical features are CMs and AVMs/AVFs, the broader phenotypic spectrum of RASA1-related disorders is still being defined. Here, we report the clinical and molecular findings of 69 unrelated individuals who had a RASA1 variant identified at ARUP Laboratories using various clinical testing methodologies. The clinical and molecular findings of several cases are described in detail to expand the current understanding of this rare vascular anomaly syndrome, and help inform the molecular testing algorithm for patients with vascular phenotypes.
Materials and methods
Subjects
All samples submitted for RASA1 testing at ARUP Laboratories within the last eight years were included in the study: 281 unrelated patients (139 males; 142 females). Cases tested positive for RASA1 variants are shown in Table 1. ARUP is a medium-sized reference laboratory that receives samples from throughout the United States of America. Data on the clinical phenotypes of patients were collected from a RASA1 specific, vascular malformation panel, or HHT panel patient history form (Supplemental Figure 1) on which the ordering physician was asked to specify the type, location, and number of vascular malformations, as well as other findings. In some cases, ARUP genetic counselors contacted the ordering clinician to clarify reported findings or obtain more information. This study was approved by the University of Utah Institutional Review Board (IRB #7275, 35637, and 20480).
Sanger sequencing
DNA was extracted from 1 mL of blood by MagnaPure Compact (Roche) extraction following manufacturer’s instructions. RASA1 coding regions and exon-intron boundaries were PCR-amplified for all patients. Primer sequences are available upon request. Amplicon fragments were bi-directionally sequenced with universal M13 primers using the Big Dye® Terminator v3.1 cycle sequencing kit and an ABI 3730 DNA Analyzer (Life Technologies, Carlsbad, CA). Sequences were compared to the RASA1 reference sequence (NM_002890.2; NG_011650.1) using Mutation Surveyor (SoftGenetics, State College, PA, USA). Sequencing results and clinical information for each patient was entered into Progeny Clinical Software version 7 (Progeny Software, Indianapolis, IN).
Next-generation sequencing and data analysis
Genomic DNA was extracted from peripheral blood using a Gentra Puregene Blood Kit (Qiagen, Valencia, CA). Custom 120 nucleotide RNA baits were designed to specifically target the exons and exon/intron boundaries of RASA1 and either four or thirteen additional vascular malformation genes, depending on which multi-gene panel was ordered. A 14-gene vascular malformation panel and 5-gene HHT panel were used to evaluate several cases (Table 2 for list of the genes for each panel). RNA baits were tiled at 5× spacing and were in replicates of 10 to increase hybridization efficiency of the targeted region (~0.1 Mb). Genomic DNA (3 µg) was sheared to 180 bp fragments. Illumina adapters were added using the Bravo automated instrument and SureSelect XT kit reagents (Agilent Technologies, Santa Clara, CA). Adapter ligated DNA underwent hybridization with the biotinylated RNA baits for 24 h at 65 °C. Hybridized DNA targets of interest were captured using streptavidin-coated magnetic beads. DNA targets of interest were eluted and barcode/indexed after a series of washes to remove the non-targeted, unbound genome. DNA quality and quantity were assessed using the LabChip Gx (Perkin Elmer, Waltham, MA). Concentrations of indexed samples from the enrichments were determined, and samples were pooled and sequenced on a HiSeq2500 instrument (Illumina, San Diego, CA) using 2 × 100 paired-end sequencing.
Sequences were aligned to the human genome reference (hg19) sequence using the Burrows–Wheeler Alignment tool (BWA 0.5.9) with default parameters [14]. PCR duplicates were removed using the Samtools package [15], and base quality score recalibration, local realignment, and variant calling were performed using the Genome Analysis Toolkit (GaTK v1.3) [16]. Potential variants that affect function were confirmed using Sanger sequencing.
Structural variation analysis
Large exonic deletions and duplications of RASA1 were identified using the P409 RASA1 multiplex ligation-dependent probe amplification (MLPA) kit (MRC-Holland, Amsterdam, Netherlands). Several individuals were evaluated for large deletions and duplications in RASA1 as part of the vascular malformation panel using a custom designed array-comparative genomic hybridization (aCGH) for targeted genes of interest (Agilent Technologies, Santa Clara, CA).
cDNA sequencing
RNA was extracted from peripheral blood and converted into cDNA for case 48 using reverse transcriptase. Exonic primers were used to specifically amplify the region of RASA1 that had the potential splicing defect. Primer sequences are available upon request.
Results
Clinical and molecular findings of cases with a RASA1 variant
Of the 281 unrelated individuals evaluated for RASA1, 69 individuals had a RASA1 variant. Clinical and molecular findings for these cases are summarized in Table 1. All clinical findings are as reported by the physician who submitted the case for molecular analysis. All cases were evaluated for RASA1 by sequencing (Sanger and/or NGS with a 14-gene panel or 5-gene panel). Many (154 cases) were also evaluated for RASA1 large deletions or duplications by multiplex ligation-dependent probe amplification (MLPA) or aCGH. Sixty cases had a pathogenic RASA1 variant that affects function of which 29 were novel, yielding an overall positivity rate of 21.4% within this cohort. Nine cases (3.2%) had a RASA1 variant of uncertain significance (VUS). The majority (75.4%) of individuals with a RASA1 variant had a report of one or more CMs, and in nearly half (44.9%) of cases the ordering provider indicated that the patient had an AVM or AVF. For several cases, limited information was available on the details of the vascular lesions reported by the referring physician.
A pathogenic variant that affects function was identified in 60 of 281 individuals (21.4%) in our cohort (Table 1). Most RASA1 variants identified that affect function caused a frameshift, premature stop codon, or disrupted splicing, and many (29/60) were novel (Table 1). The majority of individuals with a variant that affects function were reported to have multiple capillary malformations (40/60 cases); and nearly half of individuals (28/60 cases) were reported to have an AVM or AVF with most occurring in the brain or face. Roughly half of individuals with a variant that affects function had a family history of the disorder with family members exhibiting similar clinical findings (27/60 cases), while 11 cases were reported as having an unknown family history.
RASA1 deletions in CM-AVM cases
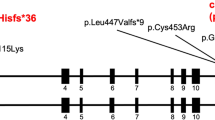
To date, only two reports of large contiguous gene deletions of RASA1 and MEF2C, a neighboring gene, in cases with 5q14.3 neurocutaneous syndrome have been published [17, 18]. In contrast, we report a relatively high prevalence (8.3%) of RASA1 exonic deletions among cases with a variant that affects function. Five cases with large RASA1 deletions of various exons were identified (Table 1 and Fig. 1). These multi-exon deletions were identified using MLPA (cases 58–60) and aCGH (cases 56 and 57). No large duplications were observed.

Large RASA1 deletions. a, b Two large RASA1 deletions detected by MLPA. c Large RASA1 deletion of the 5’UTR and exon 1 detected by aCGH
Case 59 had prior negative results by Sanger sequencing and upon re-evaluation the following year when MLPA testing became available tested positive for deletion of RASA1 exons 16–20. Surprisingly, two unrelated probands, cases 56 and 57, tested positive for the same RASA1 5’UTR and exon 1 deletion by aCGH (chr5:85,938,734-86,602,815 and chr5:86,469,478-86,613,626, respectively) (Fig. 1c). These deletions abolish the translation start site and delete exon 1 in both RASA1 isoforms 1 and 2, also known as the long (P20936-1) and short isoforms (P20936-2), respectively.
Overall clinical findings for cases with large deletions were similar to CM-AVM cases that tested positive for a deleterious RASA1 nonsense, frameshift, or splice site variant. Of the five cases with large RASA1 deletions, three presented with multiple CMs, and two presented with AVMs. One patient (case 57) was a mother with multiple CMs whose infant son died of a very large brain AVM and had no biologic material for testing. One 2-year-old patient (case 60) born with a large birthmark which covered her right face, ear, and scalp (Fig. 2a) tested negative for RASA1 by Sanger sequencing. Over the next 2 months multiple cutaneous vascular lesions developed on her trunk and limbs. Her facial lesion was later diagnosed as a cutaneous AVM, the nidus of which was found in her ear (Fig. 2a, left panel). Further testing was performed, and she was found to have a deletion of RASA1 exons 21–25 by MLPA (Fig. 1a). Another patient (case 56) with a RASA1 deletion was reported to have an AVM on the left arm and hand. Given the spectrum of possible genetic causes for this clinical presentation, this patient was assessed for genetic aberrations using a custom designed 14-gene vascular malformations NGS panel and aCGH (Table 2). Although NGS panel testing was negative, a heterozygous RASA1 deletion of the 5’UTR and exon 1 was identified by aCGH (Fig. 1c). Acknowledging the limitations of this small sample size, these clinical findings broadly match the typical clinical presentations of cases with RASA1-related disorders further emphasizing the importance of testing for large RASA1 deletions and duplications in CM-AVM cases.

Case photographs and pedigrees. a Cutaneous AVM of the ear (left panel) and large birthmark covering the right face, ear, and scalp (left and right panels) of case 60. b Pedigree of family suspicious for HHT in which exome sequencing of the proband (case 1) showed a RASA1 frameshift. c Family pedigree of case 48 with a novel RASA1 splice site variant in which the affected infant was diagnosed prenatally with cerebral AVFs. d Fetal color Doppler ultrasound demonstrates a high-flow arteriovenous shunt in the posterior fossa (affected sibling of case 48). e MRI demonstrates a signal void (arrow) representing a high flow with arteriovenous shunting along the surface of the left cerebellar hemisphere (affected sibling of case 48). f Frontal early and latter images of the left vertebral artery shows the multiple AVFs in the posterior fossa draining to a markedly dilated tributary to the left transverse sinus (affected sibling of case 48). g The thigh of case 69 had innumerable, diffuse tiny telangiectases (arrows, left panel) with significant surrounding halos giving the skin a mottled effect (left panel). Pale pink capillary malformation (~1 cm) on the left arm of case 69 (right panel)
Exome sequencing showed a RASA1 frameshift in a family suspicious for HHT
For some individuals and families with vascular anomalies such as those reported in RASA1-related disorders, the diagnostic odyssey can be quite long. Case 1 originally presented clinically with recurrent spontaneous epistaxis, what were reported as multiple dermal telangiectases, and a first-degree relative with a brain AVM and telangiectases. Having seemingly met three of four diagnostic criteria for HHT, this individual was originally diagnosed with HHT and tested for known HHT genes (ENG, ACVRL1, and SMAD4) by sequencing and deletion/duplication analysis. When no causative variant was detected in these genes, exome sequencing was pursued. Exome analysis showed a novel RASA1 frameshift variant [c.261_262del, p.(Gly89Argfs*22)], which was later found to track in additional affected family members by Sanger sequencing (Fig. 2b). Upon re-examination by the referring physician, the patient’s dermal lesions were found on the proximal extremities and trunk, rather than on the hands, mouth, and face as is typical in HHT.
NGS panel testing identifies atypical and clinically ambiguous RASA1 cases
The extensive molecular investigation of case 1 prompted the creation of a 14-gene targeted vascular malformations NGS panel (Table 2) to facilitate the accurate molecular diagnosis of patients with vascular anomalies that may be difficult to distinguish clinically or who present with atypical clinical findings. While most RASA1 variants in these cases were identified using traditional Sanger sequencing, RASA1 variants were also identified in nine cases (cases 7, 13, 23, 35, 47, 54, 55, 62, and 65) using the 14-gene vascular malformations NGS panel and two cases (cases 61 and 67) using a 5-gene HHT NGS panel (Table 2, starred genes). The descriptions of the clinical findings reported by the examining physician in many of these cases appear to suggest that they were not particularly suggestive of CM-AVM syndrome (cases 54, 55, 61, 62, 65, and 67). In particular, none of these cases were reported to have cutaneous CMs. Most had one finding that was consistent with CM-AVM syndrome, but the ordering physician had not raised the diagnosis in the clinical setting. One individual (case 54) reported to have a lung AVM and telangiectases was suspected to have HHT at the time the vascular malformation panel test was ordered. The 14-gene vascular malformations NGS panel was used to identify a nonsense variant in RASA1 [c.3028C > T, p.(Arg1010*)] in this patient that was confirmed by Sanger sequencing. The report of a lung AVM was based on a positive contrast echocardiogram (suggestive of right-to-left shunt) and an equivocal nodule on chest CT. The nodule suspected to be a lung AVM at the time molecular genetic testing was ordered, was not confirmed on a subsequent chest CT. Another individual (case 61), in which a VUS in RASA1 was detected using a 5-gene HHT NGS panel, was also reported to have a lung AVM with clinical suspicion for HHT at the time molecular genetic testing was ordered. Similarly, further investigation showed that although the patient had evidence of a pulmonary shunt, there was no lung AVM identified by chest CT.
While mucocutaneous telangiectases are more classically associated with HHT, 11 individuals with a RASA1 variant that affects function identified by NGS had dermal lesions classified as telangiectases by the ordering physician. Three of these individuals had previously tested negative for HHT causative genes (ENG, ACVRL1, and SMAD4). Dermal lesions described as telangiectases were also reported in six additional individuals (cases 10, 11, 21, 26, 27, and 45), with or without CMs or AVMs, who were found to have a RASA1 variant that affects function by Sanger sequencing (Table 1). Of note, the record of patient skin lesions relies on reports from the ordering physicians and it is not clear that all are applying the term “telangiectasia” equivalently.
Complex RASA1 case with multiple genetic findings identified by NGS
The 14-gene vascular malformations panel has a broader utility due to the ability to identify variants in multiple genes for patients with more complex clinical presentations or family history. One 16-year-old male (case 35) was diagnosed clinically with PKWS due to a leg AVM/AVF and suspected lymphangioma, in addition to cellulitis and tetralogy of Fallot. The 14-gene vascular malformations NGS panel was performed and the following variants in three separate genes were identified and confirmed by Sanger sequencing: a novel RASA1 nonsense variant [c.2269del, p.(Leu757*)], a novel PTEN splicing variant (c.1026+1 G > A), and a SMAD4 variant of uncertain significance [c.70 A > G, p.(Met24Val)] (Fig. 3a). The SMAD4 VUS was highly conserved and predicted to affect function by computational analysis algorithms (PolyPhen2, SIFT, and MutationTaster). Further investigation of the proband’s family showed that the RASA1 and PTEN variants had been maternally inherited while the SMAD4 VUS was paternally inherited (Fig. 3b). The proband’s mother did not have any clinical findings but has not fully examined; however, her sister has two children with autism spectrum disorder who tested positive for a PTEN variant and another child who died shortly after birth due to truncus arteriosus. Germline SMAD4 mutations cause a combined syndrome of HHT and juvenile polyposis that can increase the incidence of intestinal polyps [19]. Subsequent clinical examination of the proband’s father showed the presence of more than 100 colonic polyps, which were proactively assessed for malignancy as a result of this molecular diagnosis.

Complex RASA1/PTEN case identified by NGS panel testing. a Pedigree depicts which genetic variants (RASA1, PTEN, and/or SMAD4) the proband and family members carry. b NGS panel data and c Sanger sequencing data from for each variant
Novel RASA1 splice site variant in an infant prenatally diagnosed with cerebral AVFs
Next, we report a 2-year-old child (case 48) with cerebral AVF and multiple cutaneous CMs, whose mother and several family members have CMs (Fig. 2c). Sequencing analysis of the proband showed a novel synonymous RASA1 variant (c.2604G > T, p.Pro868 = ) at the 5’ end of exon 20 (Table 1). Predicted to disrupt splicing, this variant was originally classified as a variant of uncertain significance. At the time of testing, the patient’s affected mother was pregnant with a fetus whose brain AVF was detected by ultrasonography (Fig. 2d) and confirmed using fetal MRI (Fig. 2e). At birth, the newborn was neurologically normal but had mild congestive heart failure that could be controlled medically. Cerebral angiography showed a complex high flow AVF with multiple feeding arteries suspected to be the cause of the newborn’s congestive heart failure (Fig. 2f). The AVF was treated by transcatheter embolization, with subsequent resolution of the newborn’s congestive heart failure.
In order to prove pathogenicity of the novel RASA1 variant, peripheral blood was obtained from the mother and her newborn. RNA was extracted and converted into cDNA using reverse transcriptase. cDNA sequencing using exonic primers showed that the RASA1 c.2604 G > T variant disrupted splicing by causing the removal of exon 20 from the mRNA sequence (Supplemental Figure 2). Based on the family history and clinical information, we conclude that this novel RASA1 variant affects splicing. This appears to be the second report of a RASA1-associated brain AVF detected by prenatal ultrasonography and confirmed by fetal MRI as well as postnatal cerebral angiography [9].
RASA1 variants of uncertain significance
Nine cases had a variant of uncertain significance (Table 1, cases 61–69). Seven individuals had a novel or rare RASA1 missense amino acid change that was highly conserved and predicted to be damaging by one or more computational algorithms (SIFT, Polyphen, and Mutation Taster) [20,21,22]. REVEL scores are listed for each missense VUS in Table 1 [23]. Since the majority of deleterious RASA1 variants reported in CM-AVM cases have been loss-of-function mutations [13], these missense variants were classified as variants of uncertain significance. It is possible that these missense variants could cause loss of RASA1 function by disrupting splicing or altering functionally relevant protein–protein interactions.
Family concordance studies were performed for only three of nine cases with a VUS (cases 64, 68, and 69) due to limited sample availability. Case 69, who carries the p.(Val737Ile) variant, is a 14-year-old with both telangiectases and capillary malformations typical of those previously described in CM-AVM (Fig. 2g, left panel). The skin of all four limbs had innumerable, diffuse tiny telangiectases with significant surrounding halo. The halo around each telangiectasia gave the skin a mottled effect as pictured. He also had pale pink CMs ~1 cm in size on his chest (3), right shoulder (1), and left arm (1). A brain MRI was negative for AVM/AVF. Neither parent had suspicious cutaneous lesions, but two siblings had similar innumerable, haloed telangiectases; one sibling on both arms and the other sibling on hands and arms. Both siblings and their mother were found to have the same RASA1 variant identified in the patient. These combined findings suggest that the variant may be deleterious, and the mother represents non-penetrance. In case 64, the p.(Leu437Arg) variant was also identified in the affected son of the proband; although this provides further evidence for pathogenicity, the variant remains classified as uncertain as additional family members were unavailable. Because the mother of case 68 carried the p.(His695Leu) variant but has not been evaluated for internal involvement (AVMs or AVFs), and non-penetrance is possible, this variant also remains classified as uncertain.
Two cases (case 63 and 65) had a novel intronic variant of uncertain significance located near the exon (c.1254-19_1254-8del and c.1332+6T > G) that could affect splicing. Although the deletion (c.1254-19_1254-8del) for case 63 was close to the acceptor splice site of exon 9 and predicted to potentially cause a skip of exon 9 (Alamut software version 2.4, NNSPLICE and MaxEnt), the prediction algorithm scores were not definitive so the variant was classified as uncertain. The c.1332+6T > G variant in case 65 was not predicted to alter splicing (Alamut software version 2.4, NNSPLICE and NetGene2), but a neighboring nucleotide change (c.1332+5G > A) was reported in four family members with CMs [1]. This previously reported variant was also not predicted to affect splicing, so the c.1332+6T > G variant was classified as uncertain. Unfortunately, samples were not available from either family for segregation studies or splicing studies.
The majority of cases (5/9) found to have a RASA1 VUS had multiple CMs, and one of these individuals (case 68) also had a brain AVM/AVF, and thus were clinically very suspicious for CM-AVM syndrome. Two other individuals with a VUS (cases 62 and 67) had an AVM but were not reported to have CMs.
Cases that tested negative for a RASA1 variant
All other individuals in our cohort, including 127 cases reported to have multiple capillary malformations and 23 cases with a vein of Galen aneurysm or brain AVM (10 cases reported to have both), tested negative for a RASA1 variant that affects function or VUS. Compared to the clinical features of the patients with a RASA1 variant identified, the negative cohort had fewer cases with CMs (59.9% compared to 75.4% in cases with a RASA1 variant) and AVMs (33.5% compared to 44.9%). The number of cases with hypertrophy and PKWS were comparable among the cohorts. Several rare RASA1 variants were observed but were classified as benign based on their location in the RASA1 gene and/or presence in other unaffected individuals (dbSNP, ExAC, and Gnomad databases). Benign RASA1 variants and their prevalence are listed in Supplemental Table 1. One of these variants [c.1583A > G, p.(Tyr528Cys)] was originally classified as a variant of uncertain significance in a case of Southeast Asian decent because it had been reported previously in an individual with CMs [13]. It was later found to be present in ~1% of Southeast Asians (ExAC database) and was reclassified to benign.
Discussion
We report the variant detection rate among all samples received for RASA1 testing at ARUP Laboratories within the last 8 years. Of the 281 unrelated samples received for testing, 60 had a variant that affected function and nine had a variant of uncertain significance. All other cases had a benign variant or were negative for a RASA1 variant. Based on these results, our clinical mutation detection rate is 21.4% (60/281) which increases to 27% when patients without CMs were excluded (47/175). This clinical sensitivity is very similar to what we reported previously (29%) on a series of 35 consecutive cases evaluated for a RASA1 variant [12]. These rates are lower than what has been reported in other studies [1, 13]. Revencu et al. identified a RASA1 mutation in 44 (78%) of 56 probands with multifocal CMs with or without additional vascular malformations [1], and the same group identified a RASA1 mutation in 68 (68%) of 100 index patients with CM-AVM in a more recent prospective study [13]. Mutation detection frequencies in clinically ascertained cohorts differ when compared to well-characterized research study groups because patient selection criteria for molecular genetic testing are not likely to be as strict. For example, some individuals in our cohort who were referred for RASA1 testing had phenotypes that suggested other vascular conditions such as HHT, Sturge–Weber, Klippel–Trenauney syndromes, and PTEN-hamartoma tumor syndrome (PHTS), rather than only CM-AVM and PKWS. Very recently, a second form of CM-AVM (CM-AVM2) caused by germline loss-of-function mutations in EPHB4 was reported [24]. CM-AVM2 mimics RASA1-related CM-AVM and also HHT in that clinical features include multifocal CMs, telangiectases, and AVMs [24, 25]. With this new discovery, it is likely that cases with CMs and AVMs who tested negative for a RASA1 variant have an EPHB4 variant.
Over half of the RASA1 variants in our cohort were novel. Of the deleterious variants observed in our cohort, the majority caused a frameshift, premature stop codon, or disrupted splicing (Table 1). The prevalence of these loss-of-function variants suggests that haploinsufficiency is a common disease mechanism in RASA1-related disorders. It is also possible that the missense variants of uncertain significance reported here ultimately cause loss of RASA1 function by disrupting splicing or relevant protein–protein interactions. Haploinsufficiency of the Ras p21 protein activator 1 protein would cause the Ras/MAPK signaling pathway to be upregulated and disrupt the normal regulation of growth, differentiation, and proliferation of cells, likely during angiogenesis in these patients [26]. In fact, we and others have reported that somatic second hit variants in the RASA1 gene are responsible for the development of capillary malformations in CM-AVM patients [10, 13]. Mechanistically, this helps explain much of the variable expression seen in RASA1-related disorders even in individuals with the same RASA1 germline variant. Complete loss of function of RASA1 may also be responsible for the development of AVMs and AVFs in these patients, although this remains to be tested.
We also report a fairly high prevalence (8.3%) of RASA1 exonic deletions among cases that had a deleterious variant (Fig. 2). This demonstrates the importance of including deletion/duplication testing within RASA1 investigation algorithms. Of the five patients found to have a large deletion, two presented with dermal AVMs and three presented clinically with multiple CMs, one of whom had a son with a very large brain AVM (case 57). Recognizing the limitation of this small sample size, the clinical presentation of these cases largely aligned with those found in patients with other RASA1 variants that affect function. These results demonstrate that deletions represent another important mechanism of disease in individuals with CM-AVM syndrome and highlight the fact that individuals with CM-AVM syndrome who tested negative for a RASA1 variant by sequencing alone may now benefit from deletion/duplication investigation. To this end, we recommend that RASA1 deletion/duplication analysis by MLPA or other methods be performed with sequencing analysis for individuals with symptoms consistent with CM-AVM syndrome. Individuals with phenotypes consistent with CM-AVM syndrome who had molecular genetic testing by RASA1 sequencing alone in the past, in whom no variant was detected, should be considered at the time of follow-up consultation for additional deletion/duplication testing using MLPA.
Consistent with previous reports, the presence of multiple CMs in our cohort remains the most common feature associated with RASA1 mutations [12, 13]. Of the 69 individuals found to have a RASA1 variant, 52 individuals (75%) were reported to have one or more CMs. Forty-five cases (65%) with a RASA1 variant had CMs that were described as multiple. These results are consistent with a previous report of 68 CM-AVM syndrome families in which the authors concluded that mutations in RASA1 underscore the specific CM-AVM syndrome phenotype and the clinical diagnosis is based on identifying the characteristic multifocal cutaneous CMs [13]. Yet, it is important to note that not all RASA1 positive cases from our cohort had CMs that had been recognized and reported by their physician, or any reported dermal findings (cases 2, 3, 35, 42, 43, and 56). In each of these cases, an AVM was reported and was likely the main clinical feature to order RASA1 testing. Our data along with published reports highlight the importance of clinical and penetrance variability. We recommend parental testing anytime a RASA1 variant is found in the proband, since negative clinical findings on outward physical examination do not always assure negative molecular results in parents or family members.
In addition to multiple capillary malformations, patients that tested positive for a RASA1 variant frequently had an AVM and/or AVF. Of the 60 patients with a RASA1 variant that affects function detected, 27 (45%) were reported to have an AVM and/or an AVF. It is unclear how many individuals not known to have an AVM at the time of molecular genetic testing had imaging after the identification of a RASA1 variant in which an AVM/AVF was detected. Brain AVM/AVFs were the most common in our positive cohort (12 affected individuals), followed by AVM/AVFs of the face (5 individuals) and extremities (5 individuals). Consistent with previous reports, the majority of individuals with AVM/AVFs of the extremities had multiple arteriovenous microfistulas throughout the upper or lower extremity as opposed to a single, large AVM [13]. Nine cases also had hypertrophy, mainly occurring in the extremities. The percentage of patients in our study reported to have PKWS is lower (6.4%) than in a previous study performed by Revencu et al. in which 16 of 56 probands (~29%) had PKWS and greater than 80% of these patients (13 of 16) had a RASA1 mutation [1]. Otherwise, the clinical findings in our cohort broadly match the phenotypic findings from previous reports [1, 13]. However, our data is limited as it relies on the ordering physician providing the quality of clinical data.
We demonstrate the clinical utility of a custom 14-gene next-generation sequencing (NGS) vascular malformations panel (Table 2), which is useful for those patients who present with atypical clinical findings or vascular lesions that are difficult to distinguish clinically; or apparently non-syndromic vascular malformations. It is of note when considering the diagnosis of vascular malformation syndromes that the clinical presentation is extremely variable. Thus, a clinical presentation that appears upon initial evaluation to be non-familial and non-syndromic, might actually be both. A lower proportion of cases that tested positive for a RASA1 variant by NGS panel testing reported capillary malformations (5/13), than when RASA1 gene testing alone was ordered. This supports the use of NGS when the phenotype is relatively non-specific.
Several cases evaluated using this NGS diagnostic panel had reported clinical features such as “lung AVMs” and/or dermal lesions described as “telangiectasia” thus raising clinical suspicion of HHT. It is possible that the dermal lesions in these patients were actually more typical for CM-AVM syndrome. Our data suggest that although the dermal lesions of HHT and CM-AVM syndrome are described as being distinct in detailed reports of each disorder to date, many clinicians find them difficult to distinguish. And there is clearly confusion, or lack of consistency, in the use of the terms “capillary malformation” and “telangiectasia” by the ordering physicians. The distinction from telangiectases in HHT is that those seen in HHT occur overwhelmingly in the oral cavity and on the lips, hands, and face, as well as fingers and palms, are not haloed, and are finite (countable—often less than a dozen total) in number [27]. Our results suggest that patients who have tested negative for the known HHT genes may actually have a RASA1 variant, and that an NGS panel test that includes the gene associated with HHT in addition to RASA1 (i.e., ENG, ACVRL1, SMAD4, GDF2, and RASA1) may be beneficial for patients who are suspicious for HHT but do not meet at least three of four clinical diagnostic criteria for HHT (epistaxis, telangiectases, AVM, family history) [28] or have cutaneous telangiectases that are not typical for HHT. Although the appearance and location of the cutaneous capillary malformations or telangiectases described in RASA1-related disorder/CM-AVM syndrome are likely distinguishable from those in HHT for the majority of cases, there is enough similarity that making the distinction can be difficult for clinicians without specific expertise in the identification of vascular malformations. In this setting, and for patients presenting with more atypical or ambiguous phenotypic findings, a customized NGS panel and aCGH testing that includes the RASA1 gene and the recently discovered EPHB4 gene among additional vascular genes may be beneficial.
Further highlighting the clinical utility of a 14-gene vascular malformations NGS panel (Table 2), one complex case (case 35) diagnosed with Parkes Weber with an AVM/AVF of the leg with probable lymphangioma had a variant that affects function in both RASA1 and PTEN and a variant of uncertain significance in SMAD4 (Fig. 3). Panel testing was very useful in this case because other disorders featuring vascular malformations such as PTEN hamartoma tumor syndrome (PHTS) can be in the clinical differential of RASA1-related disorders. Importantly, the molecular results were useful in guiding the proper management and care of this complex vascular case and his family members (Fig. 3a). To our knowledge, this is the first report of a complex case with a deleterious variant in both RASA1 and PTEN.
Of note, most patients submitted for RASA1 testing (~75%) were negative for a RASA1 variant. Yet, nearly half of all cases (127/281) were not evaluated for a large RASA1 deletion or duplication. Such a high negative rate in patients who have multiple capillary malformations, some also with clinical findings such as a vein of Galen aneurysm or a brain AVM, suggests that further evaluation of the RASA1 gene for large deletions/duplications is warranted. Also, with the recent discovery of a new CM-AVM syndrome gene [24], cases that have previously tested negative for RASA1 should now be evaluated for variants in EPHB4. Due to the extensive phenotypic overlap between the two syndromes, it is likely that a significant proportion of cases that have tested negative for RASA1 will have an EPHB4 variant. More EPHB4 positive cases will help to define the similarities and differences between RASA1-related disorders and CM-AVM2 further.
Conclusion
We report the clinical and molecular findings of 69 unrelated cases that had a RASA1 variant among all samples sent for RASA1 testing at a clinical reference laboratory over the last 8 years. Of 281 unrelated individuals evaluated for a RASA1 variant, 60 had a variant that affects function of which 29 were novel, and 9 had a variant classified as uncertain. The publically available RASA1 mutation database (http://www.arup.utah.edu/database/RASA1/RASA1_display.php) has been updated to include these variants. Importantly, we report the first RASA1 deletions observed in CM-AVM cases at a fairly high prevalence (8.3%) among positive cases and demonstrate the clinical utility of adding deletion/duplication testing to molecular testing algorithms for patients with RASA1-related disorders. Overall, our clinical mutation detection rate is 21.4% (60/281) which increases to 27% (47/175) if patients without capillary malformations are excluded. Most individuals with a RASA1 variant had multiple CMs, and nearly half had an AVM/AVF. Our results highlight the importance of screening for large RASA1 deletions and duplications in this disorder and demonstrate the benefit of NGS testing in the molecular diagnosis of cases with complex vascular phenotypes.
References
Revencu N, Boon LM, Mulliken JB, et al. Parkes Weber syndrome, vein of Galen aneurysmal malformation, and other fast-flow vascular anomalies are caused by RASA1 mutations. Hum Mutat. 2008;29:959–65.
Eerola I, Boon LM, Mulliken JB, et al. Capillary malformation-arteriovenous malformation, a new clinical and genetic disorder caused by RASA1 mutations. Am J Hum Genet. 2003;73:1240–9.
Boon LM, Mulliken JB, Vikkula M. RASA1: variable phenotype with capillary and arteriovenous malformations. Curr Opin Genet Dev. 2005;15:265–9.
Hershkovitz D, Bercovich D, Sprecher E, Lapidot M. RASA1 mutations may cause hereditary capillary malformations without arteriovenous malformations. Br J Dermatol. 2008;158:1035–40.
Larralde M, Abad M, Luna P, Hoffner M. Capillary malformation-arteriovenous malformation: a clinical review of 45 patients. Int J Dermatol. 2014;53:458–61.
Boon LM, Revencu N, Vikkula M. Capillary malformation-arteriovenous malformation and RASA1 mutations. Chichester: John Wiley & Sons Ltd; 2011.
Thiex R, Mulliken JB, Revencu N, et al. A novel association between RASA1 mutations and spinal arteriovenous anomalies. AJNR Am J Neuroradiol. 2010;31:775–9.
Bayrak-Toydemir P, Stevenson D. RASA1 related disorders. In: Pagon RA, Bird TD, Dolan CR, Stephens K, editors. GeneReviews. Seattle: University of Washington; 2011.
Lacalm A, Fichez A, Broussin B, Abel C, Lacombe D, Guibaud L. Prenatal diagnosis of cerebral and extra-cerebral high-flow lesion revealing unknown familial CM-AVM syndrome. Ultrasound Obstet Gynecol. 2017;51:409–11. https://doi.org/10.1002/uog.17460.
Macmurdo CF, Wooderchak-Donahue W, Bayrak-Toydemir P, et al. RASA1 somatic mutation and variable expressivity in Capillary Malformation/Arteriovenous Malformation (CM/AVM) Syndrome. Am J Med Genet A. 2015;170:1450–4.
Hernandez F, Huether R, Carter L, et al. Mutations in RASA1 and GDF2 identified in patients with clinical features of hereditary hemorrhagic telangiectasia. Hum Genome Var. 2015;2:15040.
Wooderchak-Donahue W, Stevenson DA, McDonald J, Grimmer JF, Gedge F, Bayrak-Toydemir P. RASA1 analysis: clinical and molecular findings in a series of consecutive cases. Eur J Med Genet. 2012;55:91–95.
Revencu N, Boon L, Mendola A, et al. RASA1 mutations and associated phenotypes in 68 families with capillary malformation-arteriovenous malformation. Hum Mut. 2013;34:1632–41.
Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60.
Li H, Ruan J, Durbin R. Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res. 2008;18:1851–8.
McKenna A, Hanna M, Banks E, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–303.
Carr CW, Zimmerman HH, Martin CL, Vikkula M, Byrd AC, Abdul-Rahman OA. 5q14.3 neurocutaneous syndrome: a novel continguous gene syndrome caused by simultaneous deletion of RASA1 and MEF2C. Am J Med Genet A. 2011;155A:1640–5.
Ilari R, Agosta G, Bacino C. 5q14.3 deletion neurocutaneous syndrome: contiguous gene syndrome caused by simultaneous deletion of RASA1 and MEF2C: A progressive disease. Am J Med Genet A. 2016;170:688–93.
Sayed MG, Ahmed AF, Ringold JR, et al. Germline SMAD4 or BMPR1A mutations and phenotype of juvenile polyposis. Ann Surg Oncol. 2002;9:901–6.
Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4:1073–81.
Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–9.
Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods. 2014;11:361–2.
Ioannidis NM, Rothstein JH, Pejaver V, et al. REVEL: an ensemble method for predicting the pathogenicity of rare missense variants. Am J Hum Genet. 2016;99:877–85.
Amyere M, Revencu N, Helaers R, et al. Germline loss-of-function mutations in EPHB4 cause a second form of capillary malformation-arteriovenous malformation (CM-AVM2) deregulating RAS-MAPK signaling. Circulation. 2017;136:1037–48. https://doi.org/10.1161/CIRCULATIONAHA.116.026886.
Yu J, Streicher JL, Medne L, Krantz ID, Yan AC. EPHB4 mutation implicated in capillary-malformation-arteriovenous malformation syndrome: A case report. Pediatr Dermatol. 2017;34:e227–e230. https://doi.org/10.1111/pde.13208.
McCormick F. Ras GTPase activating protein: signal transmitter and signal terminator. Cell. 1989;56:5–8.
McDonald J, Bayrak-Toydemir P, Pyeritz R. Hereditary hemorrhagic telangiectasia: an overview of diagnosis, management, and pathogenesis. Genet Med. 2011;13:607–15.
Shovlin C, Guttmacher AE, Buscarini E, et al. Diagnostic criteria for hereditary hemorrhagic telangiectasia (Rendu-Osler-Weber syndrome). Am J Med Genet. 2000;91:66–67.
Acknowledgements
We thank members of the ARUP Molecular Genetics and Genomics Clinical Laboratories for assisting in the sequence analysis of these patients. We thank the ARUP Institute for Clinical and Experimental Pathology for funding this work.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Wooderchak-Donahue, W.L., Johnson, P., McDonald, J. et al. Expanding the clinical and molecular findings in RASA1 capillary malformation-arteriovenous malformation. Eur J Hum Genet 26, 1521–1536 (2018). https://doi.org/10.1038/s41431-018-0196-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41431-018-0196-1
This article is cited by
-
Genetics of brain arteriovenous malformations and cerebral cavernous malformations
Journal of Human Genetics (2023)
-
Mutation of key signaling regulators of cerebrovascular development in vein of Galen malformations
Nature Communications (2023)
-
Comprehensive targeted next-generation sequencing in patients with slow-flow vascular malformations
Journal of Human Genetics (2022)
-
Vascular anomalies: clinical perspectives
Pediatric Radiology (2022)
-
Unilateral and segmental distribution of facial erythema: is it a real port-wine stain?
Hereditas (2020)
